


Abstract
With a short particle range and high linear energy transfer, α-emitting radionuclides demonstrate high cell-killing efficiencies. Even with the existence of numerous radionuclides that decay by α-particle emission, only a few of these can reasonably be exploited for therapeutic purposes. Factors including radioisotope availability and physical characteristics (e.g., half-life) can limit their widespread dissemination. The first part of this review will explore the diversity, basic radiochemistry, restrictions, and hurdles of α-emitters.
Radionuclide strategies for curative therapy, disease control, or palliation are positioned to constitute a major portion of nuclear medicine. The range of available therapeutic radioisotopes, including α, β, or Auger electron emission, has considerably expanded over the last century (1). Matching the particle decay pathway, effective range, and relative biological effectiveness to tumor mass, size, radiosensitivity, and heterogeneity is the primary consideration for maximizing therapeutic efficacy. β-emitting radioisotopes have the longest particle pathlength (≤12 mm) and lowest linear energy transfer (LET) (∼0.2 keV/μm), supporting their effectiveness in medium to large tumors (Fig. 1). Although the long β-particle range is advantageous in evenly distributing radiation dose in heterogeneous tumors, it can also result in the irradiation of healthy tissue surrounding the tumor site. Conversely, Auger electrons have high LET (4–26 keV/μm) but a limited pathlength of 2–500 nm that restricts their efficacy to single cells, thus requiring the radionuclide to cross the cell membrane and reach the nucleus. Finally, α-particles have a moderate pathlength (50–100 μm) and high LET (80 keV/μm) that render them especially suitable for small neoplasms or micrometastases. A recent clinical study highlighted the ability of α-radiotherapy to overcome treatment resistance to β-particle therapy, prompting a paradigm shift in the approach toward radionuclide therapy (2).

Comparison of therapeutic particle energies, particle ranges, LET, and DNA damage potencies.
For optimized therapeutic efficacy, the α-cytotoxic payload is expected to accumulate selectively in diseased tissue and deliver a sufficient radiation dose to tumor sites while sparing normal organs and surrounding healthy tissue. Some α-emitting radionuclides (e.g., radium dichloride) demonstrate intrinsic bone-targeting properties, but most radionuclides require conjugation to carrier molecules for specific delivery to tumor cells. Targeted α-therapy relies on the significant differential targeting properties of a molecular vector in delivering the lethal α-payload to cells expressing higher target concentrations. Consequently, α-emitting radionuclides have been conjugated to a wide range of biomolecules, antibodies, peptides, small-molecule inhibitors, and nanocarriers. Numerous α-conjugates showing promising preclinical outcomes are now being evaluated in clinical trials or salvage therapy studies.
α-EMITTING ISOTOPE RADIOCHEMISTRY
The α-particle is a naked 4He nucleus with a +2 charge; its extreme mass compared with that of electrons suppresses deflection of the particle, and its track is almost linear. α-particles are monoenergetic, with initial kinetic energy of between 5 and 9 MeV, yielding a corresponding particle range of 50–100 μm (Fig. 1). α-particles are effective ionizing agents and are classified as high LET. Because α-particles cannot be directly imaged in vivo, the γ-photons, characteristic x-rays, or bremsstrahlung radiation that accompany decay of the parent radionuclide are often used for quantifying target uptake, dosimetry, and therapy response.
Complex molecular pathways are initiated when α-particles interact with biologic tissue (3). The primary target of high-LET radiation is DNA, and a single α-particle track can result in irreparable double-strand breaks (4). Nucleus traversal by α-tracks correlates with cytotoxicity, whereas traversal through the cytoplasm results in more moderate radiation-induced effects (4,5). In contrast, β-particle irradiation produces mainly single-strand breaks, exhibiting approximately 500 times lower cytotoxic potency than α-particles (Fig. 1) (3). The cross-fire effect is the ability of a particle to induce damage to multiple neighboring cells, offering an advantage in heterogeneous tumors (Fig. 2). Because of the particle range, this cross-fire effect is thought to be higher with β-emitters, but recent studies showing α-particles to have a significant therapeutic effect on large tumors question this concept (6–8). In addition to direct effects, indirect radiation effects have been observed. The radiation-induced bystander effect—DNA damage in cells surrounding irradiated cells but not directly exposed to radiation—also contributes to the impact of α-radiation (6). The mechanism of this effect is not fully understood but is hypothesized to result from extracellular reactive oxygenated species, chromosomal instabilities, or other abnormalities. Finally, the abscopal effect, resulting from a radiation-induced immune response, is characterized by a therapeutic response in remote lesions (9). Importantly, compared with β-particle radiotherapy, which relies mainly on the formation of reactive oxygen species, the cell-killing efficiency of α-particles was shown to be independent of cellular oxygenation (10).

Indirect mechanisms increasing α-particle lethal potency, including cross-fire effect (CF), radiation-induced bystander effect (RIBE), and abscopal effect (AbsE) (6).
Because of the different types of biologic damage caused by high and low LET, one should consider the relative biological effectiveness factor when performing dosimetry calculations so that the estimated absorbed dose reflects the probability and relative severity of a biological effect (11). Based on in vitro experiments, if the chosen endpoint is deterministic (e.g., therapeutic efficacy or toxicity), the relative biological effectiveness ranges from 3 to 7 and should be used when predicting the benefit of α-therapy. If the endpoint is stochastic, such as cancer induction, the relative biological effectiveness for α-particles is approximately 20 (11). Human experience, however, has indicated lower toxicity than expected and highlights the dire need to develop accurate dosimetry measurement techniques for α-emitters.
α-emitting radionuclides with potential applications for radiotherapy are presented below. Because most α-emitters are progeny in a common decay chain (or family)—either direct progeny or separated by short-half-life (t½) radioactive intermediates—we elected to present radioisotopes of the same family together. Radioactive decay through multiple radioactive progeny is referred to as the in vivo generator or nanogenerator approach (12). This approach offers the significant advantage of enhancing toxicity by delivering several cytotoxic radionuclides to the tumor but also conversely suffers from the major hurdle of progeny redistribution.
211At
211At can be cyclotron-produced by bombarding natural bismuth with a medium-energy α-particle beam (28–29.5 MeV) using the 209Bi(α,2n)211At reaction (13). Even though the production and purification of 211At are inexpensive, the number of accelerators capable of generating a 28-MeV α-particle beam limits the availability of this isotope (13).
With a t½ of 7.2 h, 211At decays via a branched pathway to stable 207Pb, emitting α-particles via 2 pathways (Table 1). 211At emits K x-rays with its α-decay to 211Po, allowing for sample counting and scintigraphic imaging of 211At in vivo (14). Astatine belongs to the halogen family, and radiolabeling can be performed by adapting radioiodination chemistry (15). Tin precursors and prosthetic groups have been used to label small molecules, peptides, or antibodies (15). The carbon–astatine bond is relatively weak, and the release of free astatine can result in undesired toxicity (16). Similar to iodine, free astatine accumulates in the thyroid, stomach, and macrophage-bearing organs such as the spleen and lung.
α-Emitters for Radiotherapy, Together with Their Decay Progeny
225Ac/213Bi
The main source of 225Ac is currently 229Th generators (t½ = 7.3 y), which can be milked over a 3-wk period and allow the separation of 225Ra and 225Ac (17). The Oak Ridge National Laboratory 299Th generator produces up to 33.3 GBq per year. However, because of the limited number of generators worldwide, there is a severe shortage of this isotope for preclinical and clinical research. The 225Ac shortage also inhibits 225Ac/213Bi generator manufacturing (18).
Possible pathways toward increasing 225Ac production include high-energy proton spallation of 232Th. A triinstitutional collaboration among Oak Ridge, Brookhaven, and Los Alamos National Laboratories recently produced millicurie quantities of 225Ac by irradiating a natural thorium target at beam energies of between 78 and 192 MeV (19). Using this method, a 10-d irradiation campaign of a 5 g/cm2 thorium target was able to produce curie levels of 225Ac (19). The quality of the accelerator-produced 225Ac was equal to that of the 229Th generated; however, the impact of coproduced 227Ac remains to be evaluated (19).
225Ac (t½ = 10.0 d; 5.8-MeV α-particle) decays sequentially through 6 dominant daughters to stable 209Bi (Table 1). Decay of a single 225Ac atom yields 4 net α-disintegrations and 3 β-disintegrations together with the emission of 2 useful γ-emissions; it is therefore classified as a nanogenerator (12). The 225Ac daughter 213Bi (t½ = 45.6 min; 97.8% β−, 2.2% 6-MeV α-particle) is a widely studied radionuclide for targeted α-therapy in preclinical and clinical studies. 213Bi forms stable complexes with nitrogen-rich chelators such as CHX-A″-DTPA (2-[p-isothiocyanatobenzyl]-cyclohexyldiethylenetriaminepentaaceticacid) or NETA ({4-[2-(bis-carboxymethylamino)-ethyl]-7-carboxymethyl-[1,4,7]triazonan-1-yl}-aceticacid), and both 213Bi and 225Ac are stable on coordination by the DOTA chelator (20). Free 225Ac-acetate accumulates primarily in the liver and bone (percentage injected dose per gram: 111.8 ± 2.13 and 9.15 ± 1.2, respectively) (21). However, once chelated by DOTA, both liver uptake and bone uptake are significantly reduced (to 1.29 ± 0.25 and 0.98 ± 0.10, respectively) (21). The 225Ac daughters 221Fr and 213Bi will preferentially accumulate in the kidneys and urine.
227Th/223Ra
227Th and 223Ra are both available on separation from their mutual parent, 227Ac (t½ = 21.7 d) (22). Clinical production of 223Ra uses 227Ac/227Th-based generators (23). Parent isotopes are loaded on actinide chromatographic resin, and 223Ra-chloride solution is obtained after elution with 1 M HCl or HNO3, subsequent purification on a cation exchange column, evaporation, and dissolution in saline solution (24).
227Th (t½ = 18.7 d; 6.0-MeV α-particle) and its daughter, 223Ra (t½ = 11.4 d; 5.7-MeV α-particle), act as nanogenerators, releasing up to 4 high-energy α-particles before reaching stable 207Pb (Table 1). Emission of γ-photons allows for scintigraphic imaging of both isotopes. Biodistribution of 227Th-citrate indicates high uptake in the femur and parietal bone (25). 223Ra is an alkaline earth metal similar to calcium that, like 227Th, preferentially accumulates in sites of bone mineralization, binding into hydroxyapatite. γ-ray spectroscopy of the femur showed that, if released, 223Ra redistributes to the bone because of the α-recoil energy, resulting in an increased dose to the bone surface (25). The lack of suitable chelating agents to coordinate 223Ra limits the development of radioconjugates. On the other hand, 227Th with its +4 oxidation state can be stably chelated by DOTA (26) and octadentate chelator with hydroxypyridinone coordinating moieties (e.g., N-methyl-3,2-hydroxypyridinone [Me-3,2-HOPO]) (27).
224Ra/212Bi
224Ra, 212Pb, and 212Bi are produced by generators loaded with their long-lived parent, 228Th (28). Severe radiolytic damage to the resin of the 228Th-based generators was observed, and they were replaced by 224Ra-based generators, from which 212Bi and 212Pb are obtained selectively (29).
224Ra (t½ = 3.6 d; 5.7-MeV α-particle; 241-keV γ-particle) decays into stable 208Pb, producing 4 net α-particles and 2 β-particles, with the main recoil daughters being 212Pb (t½ = 10.6 h; 93.5-keV β−-particle) and 212Bi (t½ = 60.6 min; 36% 6.1-MeV α-particle) (Table 1). Because of its bone-seeking properties, 224Ra was initially used to treat ankylosing spondylitis (30). Even though 212Pb decays via a β-emission, its increased t½, as compared with 212Bi, allows for delivery of up to 10 times more dose per unit of administered activity, together with dose preparation and administration that are more routine. Although 212Pb forms a stable complex with the DOTA chelator, acid-catalyzed dissociation was reported. The TCMC chelator (1,4,7,10-tetrakis(carbamoylmethyl)-1,4,7,10-tetraazacyclododecane) was later developed and demonstrated extremely high stability for the Pb(II) ion (31). During 212Pb decay, γ-ray emissions compete with internal conversion over 30% of the time. The ejection of conversion electrons brings 212Bi to highly ionized states (e.g., Bi5+ and Bi7+), destabilizing the bismuth complexes and ultimately liberating the radionuclide (32). Although free 212Pb accumulates in the blood, liver, bone, and kidneys, 212Bi accumulates mainly in the kidneys and urine.
APPROACHES AND SPECIAL CONSIDERATIONS FOR HANDLING AND ADMINISTERING α-EMITTERS
Because an α-particle of at least 7.5 MeV is required to penetrate the protective skin layer (0.07 mm thick), pure α-emitters do not constitute an external radiation hazard. The main concern is internalization and energy deposition in healthy living tissues (33). Untoward radiation effects to humans from α-exposure include cancer induction, genetic diseases, teratogenesis, and degenerative changes; the respiratory tract, bone, liver, and reticuloendothelium system are the most important target tissues (33). The tumorigenesis potential of α-radiation was demonstrated after irradiation of human benign prostate epithelial cells in mice (34). Moreover, because of the bystander mutagenic effect, mutations and chromosomal aberrations have been observed in the DNA of cells that received no direct α-particle exposure, indicating that the current genotoxic risks of α-emitters are underestimated (35).
Proper handling of α-emitters is radionuclide-dependent, and each progeny must be considered because periodicity changes with decay. In the handling of α-emitters, special equipment to detect α-particles, such as ZnS(Ag) scintillators, should be available in addition to Geiger–Mueller survey meters (36). Allowable removable contamination levels for α-emitters are about 10 times lower (3.3 Bq/100 cm2) than for β-emitting radionuclides. A well-ventilated hood or, ideally, a glove box should be used in the handling of α-emitters with low abundance and low-energy γ-emission. If highly energetic γ-rays are emitted during the radionuclide decay, all work should be performed in a shielded hot cell or behind 15-cm (6-in) lead bricks using manipulator arms or remote-handling conditions (29). Extra precautions, such as trapping or gas-tight enclosures, should be considered when volatile daughters such as radon are emitted. Double gloving is recommended. Wipe tests should be performed and monitored with a γ-counter and a liquid scintillation counter.
For clinical production, centralized production should be considered for isotopes of appropriate t½. Radiochemists should be trained and have access to working and waste storage areas designed for a dedicated α-emitting isotope radiochemistry. Clinical doses should be prepared and injected once secular equilibrium is reached. Special considerations for 223RaCl2 preparation, administration, and patient release were reported for a phase I clinical study to evaluate ascending doses of 223RaCl2 at Memorial Sloan Kettering (37).
TARGETED α-THERAPY: VECTORS AND RADIOLABELING TECHNIQUES
Targeting moieties for targeted α-therapy include antibodies, peptides, or small molecules; each possesses advantages and pitfalls. Compared with small molecules, antibodies show favorable biodistribution, with high tumor uptake and low accumulation in healthy tissues. One notable example is prostate-specific membrane antigen small-molecule inhibitors (2); accumulation of these small-molecule drugs in the salivary glands is high, whereas the prostate-specific membrane antigen-specific antibody J591 shows low uptake (38). However, the longer blood circulation time of the antibody increases the risk of hemato- and myelotoxicity (6). On the other hand, small molecules and peptides exhibit higher tumor penetration and faster clearance (6). Because targeting moieties have a broad spectrum of pharmacokinetic profiles, it is important to match the physical t½ of the therapeutic radionuclide to the biologic t½ of the vector.
A radionuclide is conjugated to its vector using either a prosthetic group (211At) or a chelate (227Th, 225Ac, 212Pb, 213/212Bi). Vectors should be functionalized before the radiolabeling, and 1-step radiolabeling is preferred, especially with short-t½ radionuclides. However, the development of α-particle radioimmunoconjugates may require more complex procedures. Radioastatination of antibodies is usually performed using a 2-step method in which an aromatic organotin precursor bearing an activated ester is radiolabeled and then conjugated to the antibody (39). Radiolabeling of DOTA conjugates with 225Ac and 227Th requires harsh conditions (high temperatures, pH extremes) that are not always compatible with sensitive biomolecules such as antibodies (26). McDevitt et al. developed a 2-step radiolabeling method in which an isothiocyanate C-functionalized derivative of the DOTA chelator is radiolabeled and then conjugated to the antibody at 37°C (40); however, this method suffers from low radiochemical yields (≤10%) because of hydrolysis of the isothiocyanate moiety. Maguire et al. later proposed a 1-step method for 225Ac radiolabeling of monoclonal antibodies that allows for radiochemical yields of up to 80% (41). Other approaches imply the development of new chelators that form stable complexes at room temperature. Ramdahl et al. reported superior properties with respect to 227Th radiolabeling and stability using Me-3,2-HOPO compared with the DOTA chelator (27).
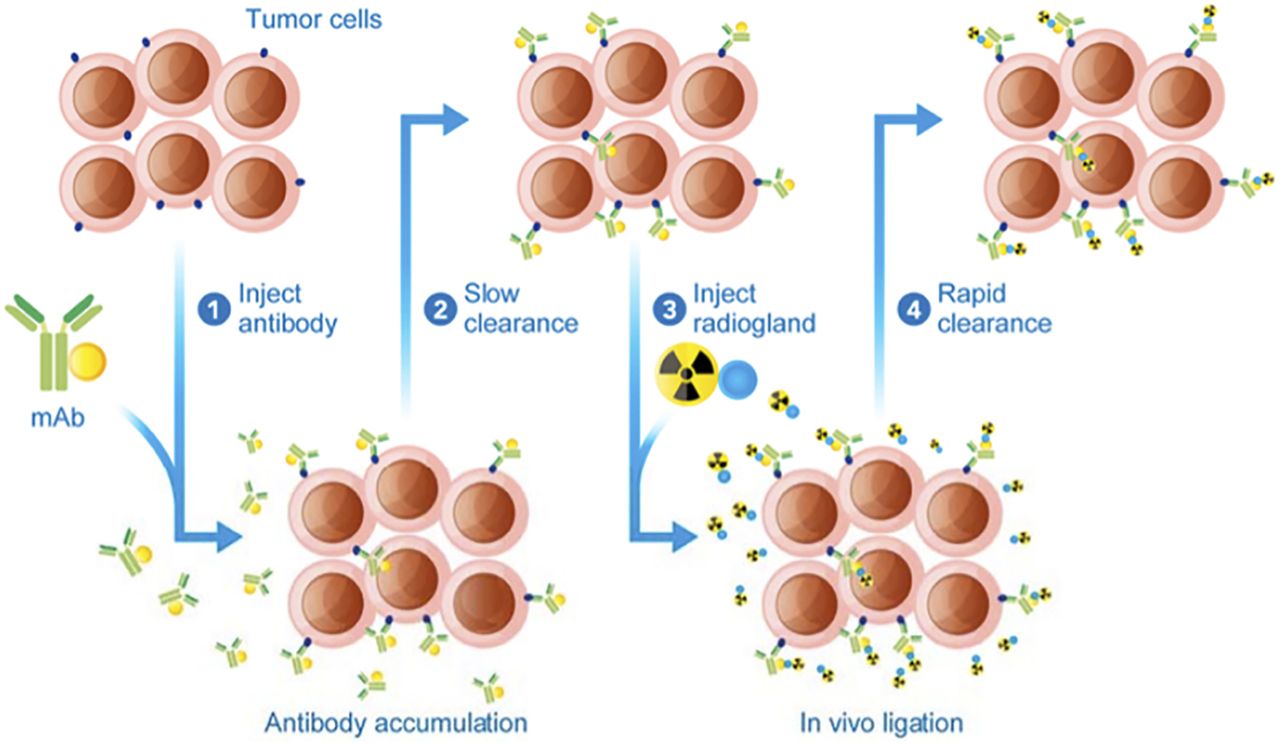
Blood toxicity and normal-tissue irradiation, caused by the slow kinetic clearance of antibodies, led to the development of an alternate delivery approach called pretargeting, which separates administration of the targeting vector from that of the radioisotope (Fig. 3) (42). First, an unlabeled antibody that binds both an antigen and the radioligand is administered, accumulates in the tumor, and slowly clears from the blood and nontargeted tissues. A low-molecular-weight radioligand is subsequently administered and diffuses into the tumor, binding to the antigen-associated pretargeting conjugate. The rapid clearance of any excess radioligand results in improved tumor–to–normal-tissue ratios and lower radiation doses to healthy organs (42). Interaction between the pretargeted antibody and the radioligand uses the extraordinarily high affinity of avidin (or streptavidin) for biotin (43), bispecific antibodies (44), or bioorthogonal chemistry (45). This approach combines the advantages of antibodies (e.g., high targeting efficiency, penetration, long residence time) with those of small molecules (rapid clearance). Moreover, this technique allows the association of antibodies with short-t½ radionuclides, such as 211At (46) or 213/212Bi (47), increasing their therapeutic potential. Applicability and efficacy in humans, though, still need to be proven, and the antibody–antigen internalization should occur either through a slow process or not at all.

Schematic representation of in vivo pretargeting (42). mAb = monoclonal antibody.
CONTROLLING THE FATE OF THE DAUGHTERS
On α-emission, recoil energy imparted to the daughter (100 keV) is about 1,000 times higher than the binding energy of any chemical bond, resulting in release of the daughter. Redistribution depends on the distance covered during the recoil process, diffusion processes, and active transport as well as the intrinsic affinity of the radionuclide for certain organs. The time to reach the target and the toxicity to healthy organs are impacted by the t½ of the daughter. Redistribution of the recoil progeny is extremely difficult to measure and is performed mostly in postmortem ex vivo analysis of organs.
Redistribution of daughters compromised the continuation of a clinical study using 224Ra; 8% of 220Rn, the gaseous 224Ra daughter, was shown to leave the body, and high uptake of 212Pb and 212Bi was observed in the red blood cells, kidneys (212Bi), and liver (212Pb) (48). On the other hand, low redistribution was demonstrated with 223Ra daughters in mice and confirmed in humans (49).
Redistribution of 213Bi to the kidneys is a main limitation of 225Ac radiotherapy. Schwartz et al. evaluated the contribution of nonequilibrium 213Bi to kidney dose in mice via γ-ray spectroscopy immediately after tissue harvest and at secular equilibrium (Fig. 4A) (50). The average absorbed dose to the kidneys was determined to be 0.77 Gy⋅kBq−1, of which 60% was attributed to nonequilibrium 213Bi excess (50).

Redistribution of α-emitter daughters: approaches to controlling their fate. (A) γ-ray spectroscopy of BALB/C mouse kidneys 96 h after injection of 225Ac-HuM195. Peaks (440 keV) indicate presence of nonequilibrium 213Bi in kidneys. (Reprinted with permission of (50).) (B) Internalization and retention of 213Bi and 221Fr daughters in vitro after binding of 225Ac-J591 in LNCaP cells. (Reprinted with permission of (12).) (C) High-resolution autoradiography evaluating spread of 224Ra progeny (212Pb) after intratumoral implantation of 224Ra wires in HCT15 tumor model in nude mice. Hematoxylin and eosin staining shows correlation with necrotic domains. (Reprinted with permission of (52).) (D) Gold-coated lanthanide phosphate nanoparticle allowing retention of 225Ac and its daughters (54). (E) Heavy-metal chelation effect on 213Bi renal uptake 24 h after injection of 225Ac radioimmunotherapy. (F) Furosemide and chlorothiazide effect on 221Fr and 212Bi renal uptake 24 h after injection of 225Ac radioimmunotherapy. %ID = percentage injected dose; DMPS = 2,3-dimercapto-1-propanesulfonic acid; DTPA = diethylenetriaminepentaacetic acid; i.p. = intraperitoneally.
The use of α-emitters with a short radioactive t½ and simple decay schemes (e.g., 213Bi or 211At) is an effective solution to daughter redistribution. Nevertheless, the higher cytotoxicity of radioisotopes with a longer t½ and decay through numerous progeny motivated the development of approaches to control the fate of the daughters. These include a high degree of nanogenerator cellular internalization. High retention of 221Fr and 213Bi inside LNCaP cells was shown in an internalization study with 225Ac-J591. Tumor samples revealed 88% retention of 221Fr and 89% of 213Bi at 225Ac secular equilibrium (Fig. 4B) (12).
A second approach relies on the development of a new form of brachytherapy, referred to as diffusing α-emitter radiation therapy. This approach, developed by Arazi et al., involves local administration of wire sources impregnated with radionuclides such as 224Ra in or near the solid tumor tissue (51). Necrotic regions of several millimeters were observed around the therapeutic source in several tumor models (Fig. 4C) (52). Autoradiography showed a larger distribution around the source for the later decay daughters, 212Bi and 212Pb, compared with the earlier decay daughters, 220Rn and 216Po. Redistribution of 212Pb to the kidneys was observed to be based on tumor size: 90% for 0.1-g tumors but only 12% for 2.4-g tumors (51).
Encapsulation of α-emitting radionuclides into nanocarriers was evaluated to retain recoil daughters at the tumor site. 223Ra encapsulation in pegylated liposomal doxorubicin demonstrated sufficient stability in vitro. Skeleton uptake remained lower than for free 223Ra, and higher uptake of the 223Ra daughters, 211Pb and 211Bi, was observed in the kidneys (53). 225Ac-doped multishell nanoparticles were evaluated to encapsulate 225Ac and its daughters (Fig. 4D) (54). Nanoparticles with 4 GdPO4 shells followed by gold coating demonstrated the greatest retention of 255Ac (>99.99%) and its daughters, with up to 98% of 221Fr retained (54).
The use of metal-chelation therapy and diuretics was investigated by Jaggi et al. to reduce renal toxicity during 225Ac radioimmunotherapy (55). Dithiols, known to chelate and enhance the urinary excretion of 213Bi, reduced the renal 213Bi activity as early as 6 h after radiotherapy (Fig. 4E) (55). An increase in 213Bi blood activity was observed in mice, but this phenomenon was not observed in cynomolgus monkeys (55). Furosemide and chlorothiazide, 2 diuretics that inhibit the tubular reabsorption of alkali metals, also significantly reduced 221Fr renal activity (Fig. 4F) (55). Though effective with long-circulating biomolecules, such an improvement might not be observed with fast-clearing molecules.
CONCLUSION
The combination of DNA double-strand breaks and indirect cytocidal effects such as cross-fire or radiation-induced bystander effects provides α-particles with exceptional cell-killing potency. Important caveats for the use of α-emitting radionuclides include production and availability limitations, together with redistribution of daughters. Solutions to these issues are currently being investigated and should allow for more widespread development of α-emitter radiotherapy. Part 2 of this educational review will explore the current preclinical and clinical uses of α-radiotherapy.
Footnotes
Learning Objectives: On successful completion of this activity, participants should be able to (1) cite α-emitter families available for therapeutic use and understand their current production limit; (2) consider radiation safety concerns when handling α-emitters; and (3) overcome radiolabeling and daughter redistribution hurdles with the approaches described in this educational review.
Financial Disclosure: This work is supported by the Radiochemistry and Molecular Imaging Probe Core, which is supported in part by NIH/NCI Cancer Center Support Grant P30 CA008748. Jason Lewis is supported by the Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and by the Center for Experimental Therapeutics of Memorial Sloan Kettering Cancer Center. Sophie Poty is supported by the François Wallace Monahan Fellowship from the JLM Benevolent Fund. The authors of this article have indicated no other relevant relationships that could be perceived as a real or apparent conflict of interest.
CME Credit: SNMMI is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to sponsor continuing education for physicians. SNMMI designates each JNM continuing education article for a maximum of 2.0 AMA PRA Category 1 Credits. Physicians should claim only credit commensurate with the extent of their participation in the activity. For CE credit, SAM, and other credit types, participants can access this activity through the SNMMI website (http://www.snmmilearningcenter.org) through June 2021.
Published online Mar. 15, 2018.
- © 2018 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication August 4, 2017.
- Accepted for publication February 3, 2018.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Mathematical Modeling Unveils Optimization Strategies for Targeted Radionuclide Therapy of Blood Cancers
- Effectiveness of 225Ac-Labeled Anti-EGFR Radioimmunoconjugate in EGFR-Positive Kirsten Rat Sarcoma Viral Oncogene and BRAF Mutant Colorectal Cancer Models
- {alpha}-Labeling of J591, an Antibody Targeting Prostate-Specific Membrane Antigen: The Technique and Considerations from the First Dedicated Production Lab at an Academic Institution in the United States
- Signaling Network Response to {alpha}-Particle-Targeted Therapy with the 225Ac-Labeled Minigastrin Analog 225Ac-PP-F11N Reveals the Radiosensitizing Potential of Histone Deacetylase Inhibitors
- {alpha}-Labeling of J591, an Antibody Targeting Prostate-Specific Membrane Antigen: The Technique and Considerations from the First Dedicated Production Lab at an Academic Institution in the United States
- Glypican-3-Targeted 227Th {alpha}-Therapy Reduces Tumor Burden in an Orthotopic Xenograft Murine Model of Hepatocellular Carcinoma
- Production and Supply of {alpha}-Particle-Emitting Radionuclides for Targeted {alpha}-Therapy
- Radiotheranostic Agent 64Cu-cyclam-RAFT-c(-RGDfK-)4 for Management of Peritoneal Metastasis in Ovarian Cancer
- Cellular and Genetic Determinants of the Sensitivity of Cancer to {alpha}-Particle Irradiation
- Leveraging Bioorthogonal Click Chemistry to Improve 225Ac-Radioimmunotherapy of Pancreatic Ductal Adenocarcinoma