


Abstract
Over the last 2 decades, significant progress has been made in our understanding of the genomics, tumor immune microenvironment, and immunogenicity of malignant melanoma. Historically, the prognosis for metastatic melanoma was poor because of limited treatment options. However, after multiple landmark clinical trials displaying the efficacy of combined BRAF/MEK inhibition for BRAF-mutant melanoma and the application of immune checkpoint inhibitors targeting the programmed death-1, cytotoxic T-lymphocyte antigen-4, and lymphocyte activation gene-3 molecules, overall survival rates have dramatically improved. The role of immune checkpoint inhibition has since expanded to the neoadjuvant and adjuvant settings with multiple regimens in routine use. Personalized therapies, including tumor-infiltrating lymphocytes that are extracted from a patient’s melanoma and eventually reinfused into the patient, and messenger RNA vaccines used to target neoantigens unique to a patient’s tumor, show promise. Improvements in accompanying imaging modalities, particularly within the field of nuclear medicine, have allowed for more accurate staging of disease and assessment of treatment response. Continued growth in the role of nuclear medicine in the evaluation of melanoma, including the incorporation of artificial intelligence into image interpretation and use of radiolabeled tracers allowing for intricate imaging of the tumor immune microenvironment, is expected in the coming years.
Malignant melanoma (hereafter melanoma) is an aggressive malignancy of pigment-producing cells called melanocytes (1). Melanocytes are found throughout the body in the skin, eyes, ears, gastrointestinal tract, and multiple mucous membranes, and thus 9 distinct subtypes of melanoma are recognized (2). Cutaneous melanoma, arising from melanocytes of the skin, accounts for the vast majority of melanoma diagnoses globally across all populations and ethnicities (3). Approximately 5% of all cases of melanoma are noncutaneous in origin; of these cases, 50% affect regions within the head and neck, with the nasal cavities and sinuses the main sites of disease (2). Uveal melanoma, the most common primary malignancy of the eye in adults, results from malignant transformation of melanocytes within the uveal tract, though it is pathophysiologically distinct from cutaneous melanoma (4). For the purposes of this review article, we are referring to cutaneous melanoma when addressing melanoma from this point onward.
The role of radiology in many areas of medicine, particularly oncology, continues to grow at a rapid pace. CT, ultrasound, and MRI play critical roles in the initial staging, treatment response assessment, and surveillance of many malignancies. The combination of 18F-FDG PET with low-dose CT, termed PET/CT, became commercially available in 2000 (5). PET/CT has resulted in significant improvements in treatment response assessment, correlation of areas of 18F-FDG uptake with underlying anatomic structures, and changes in surgical management in patients with resectable melanoma (6,7). Similarly, use of lymphoscintigraphy-assisted sentinel lymph node (LN) biopsy, initially developed as a minimally invasive technique for nodal staging in patients with melanoma, has been expanded to many solid tumor types (8,9).
EPIDEMIOLOGY
Worldwide, there were 324,635 new cases of melanoma and 57,043 deaths from melanoma in 2020 (3). In the United States, age-adjusted rates of new cases of melanoma have been rising by 1.2% on average each year from 2010 to 2019. Despite this, because of significant improvements in earlier detection of disease and treatment options, death rates have been falling by 3.3% on average each year from 2011 to 2020 (10).
Notable differences in incidence, stage at time of diagnosis, and overall survival (OS) have been observed among non-Hispanic White and Black patients within the United States. Melanoma is more common among White patients than Black patients, with incidence rates of 30.6 and 0.9 cases per 100,00 individuals, respectively, from 2016 to 2020 (10). During this period, Black patients were more likely to be diagnosed with advanced disease, with 16.9% of cases with evidence of metastases at the time of diagnosis, compared with 4.8% of cases in White patients. Differences in OS exist between these 2 patients populations as well; when stratified by race and stage, Black patients had significantly lower OS for stage I and stage III disease than White patients (11).
Multiple risk factors for the development of melanoma have been established; exposure to ultraviolet radiation through sunlight and resultant sunburns is the most common. Ultraviolet radiation exposure through both ultraviolet A and ultraviolet B light results in DNA double-strand breaks and generation of reactive oxygen species that can indirectly damage DNA (12). Other risk factors include a family history of melanoma, certain phenotypic characteristics, and use of indoor tanning beds (13,14).
STAGING OF DISEASE AT TIME OF DIAGNOSIS
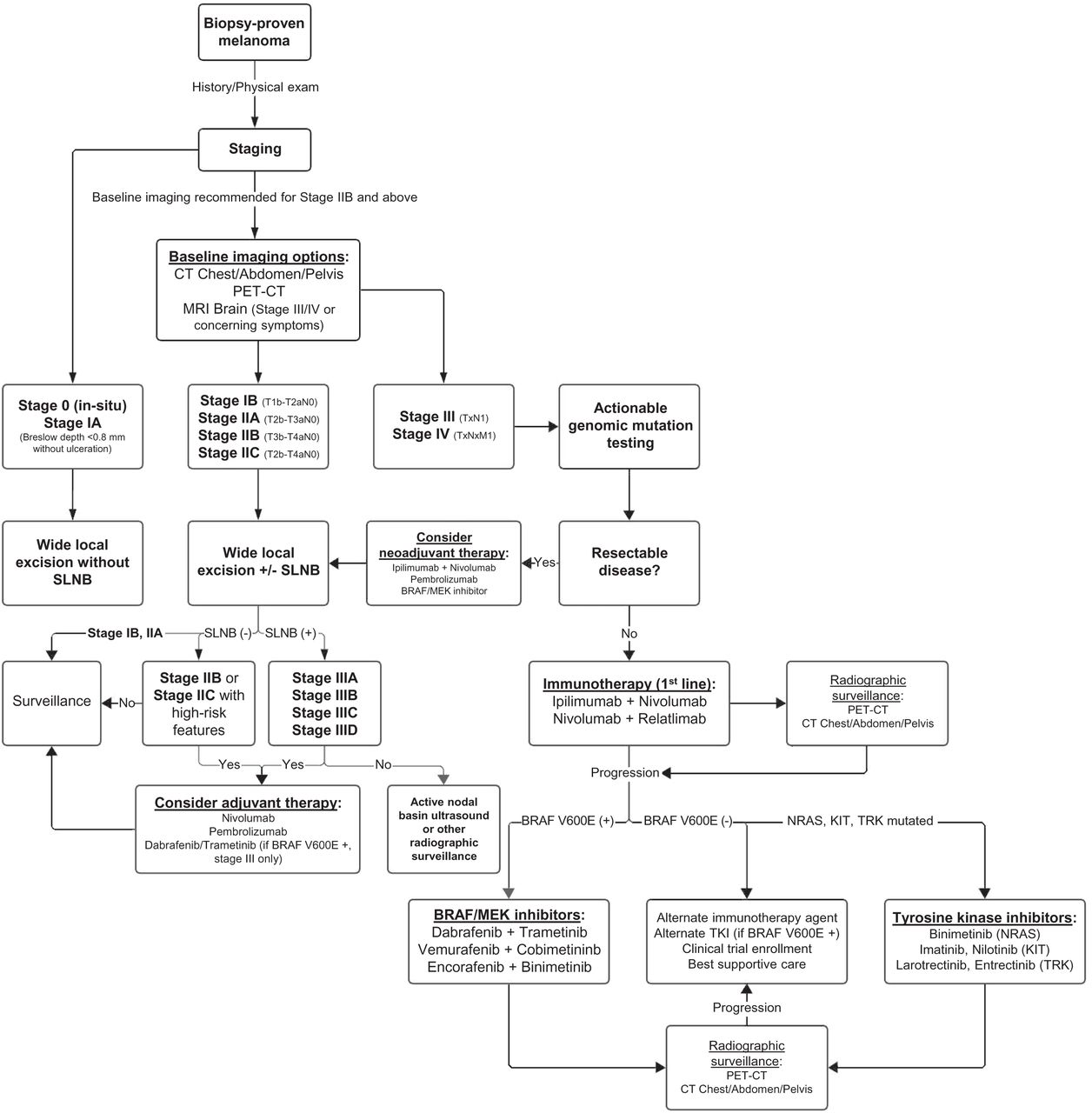
Initial staging of biopsy-proven melanoma is performed according to the American Joint Committee on Cancer eighth edition staging system; this system retains the traditionally used characterizations of the primary tumor, regional LNs, and sites of distant metastases (15). Primary tumor size is reported as tumor (Breslow) thickness in millimeters. The presence or absence of ulceration, defined as the lack of intact epithelium over the melanoma, is prognostically relevant, as patient outcomes with ulcerated primary tumors are worse (16). Regional LNs are a common site of locoregional involvement of melanoma, and assessment for nodal disease is necessary for complete staging (17). The role of sentinel LN biopsy (SLNB) is discussed further later in the article.
Multiple imaging modalities are currently available for the staging of melanoma and evaluation for distant metastatic disease. Commonly used cross-sectional imaging techniques include contrast-enhanced CT of the neck, chest, abdomen, and pelvis; whole-body PET/CT; and MRI. Not all patients require radiographic evaluation for systemic disease; neither the National Comprehensive Cancer Network nor the American Academy of Dermatology recommends cross-sectional imaging for asymptomatic patients with stage 0–IIA melanoma (18,19). 18F-FDG PET has been shown insensitive for detection of occult LN metastases in these patient populations, with a sensitivity of only 13%–21% and a false-negative rate of 79%. The low sensitivity of 18F-FDG PET in this context is thought to be due to the small size of metastatic deposits in sentinel LNs, as the sensitivity improves with increases in lesion size and is near 100% for metastases larger than 10 mm (20,21).
Cross-sectional imaging for staging is recommended in patients with stage IIB disease or higher (Fig. 1) (18). Although a multitude of imaging techniques is available, robust data clearly delineating which modality should be used in specific clinical contexts are lacking, and clinical practice is heterogeneous (22). Despite a lack of randomized clinical trial data, however, 18F-FDG PET/CT has outperformed conventional imaging with contrast-enhanced CT for both initial staging and detection of recurrent disease in systematic reviews and metaanalyses (23,24). Comparisons to whole-body MRI have been mostly equivocal, with the current high costs and limited availability of whole-body MRI generally making it a less viable alternative (23). The exception is in cerebral metastasis, for which, because of the background high uptake of glucose, PET/CT is insensitive. For this reason, MRI of the brain is recommended for patients with stage III and IV disease and in all patients with symptoms concerning for intracranial metastases (18).

Treatment algorithm for melanoma from time of diagnosis and for multiple stages of disease.
MANAGEMENT OF PRIMARY CUTANEOUS MELANOMA
After a histologic diagnosis of melanoma and subsequent clinical staging, surgical resection via wide local excision remains the mainstay of treatment for early-stage melanoma (25). Wide excision is performed down to, but not including, the muscle fascia, and the recommended width of resected margins is dependent on the Breslow thickness (19). Surgical staging of regional LNs is a critical component of prognosis and systemic treatment planning for patients with localized melanoma, although it is not required in all patients. Current guidelines recommend lymphatic mapping and SLNB for all patients with a Breslow thickness greater than 1 mm (18). SLNB can also be considered for patients with a Breslow thickness between 0.8 and 1.0 mm or less than 0.8 mm when other high-risk features are present (18,26).
For patients undergoing SLNB, preoperative lymphatic mapping via lymphoscintigraphy allows for accurate identification of sentinel LNs for dissection and sampling. Initially pioneered by Morton et al., lymphoscintigraphy uses intradermal injections of dye and radiotracer at the primary tumor site, later allowing for intraoperative identification of sentinel LNs visually and by γ-probes (8). Historically, melanoma patients with a positive SLNB underwent complete LN dissection, which was associated with increased risk of lymphedema and decreased quality of life (27,28). Complete LN dissection versus observation was subsequently examined in 2 major multicenter clinical trials. The MSLT-NRASII and DeCOG-SLT studies showed that immediate complete LN dissection did not improve distant metastasis-free survival, relapse-free survival (RFS), or melanoma-specific survival compared with surveillance with delayed complete LN dissection at recurrence (29,30). Surveillance included ultrasonographic examination of the sentinel LN basin every 4 mo during the first 2 y and every 6 mo during years 3 through 5. The role of adjuvant therapy for high-risk patients with positive SLNB and other clinical features is discussed later in this article.
MOLECULAR PROFILING AND TARGETED THERAPIES
Molecular evaluation for the presence of driver mutations is the standard of care for most patients receiving systemic therapy in any setting. The most commonly observed mutation occurs in the BRAF oncogene; since its discovery in 2002, multiple targeted therapies for the treatment of melanoma have been approved (Fig. 2) (31). BRAF is a serine-threonine kinase within the RAS-RAF-MEK-ERK pathway; approximately 50% of melanomas harbor BRAF V600 mutations resulting in constitutive activation of MEK and ERK, with BRAF V600E being the most commonly observed mutation (32). Other genomic mutations that have been identified include alterations in NRAS (∼28% of cases), NF1 (∼14% of cases), and KIT (∼15%–20% of cases of acral or mucosal melanoma) (32,33). Molecular testing can be performed on tissue obtained from the primary tumor sample, tumor-involved regional LNs, or distant metastases.

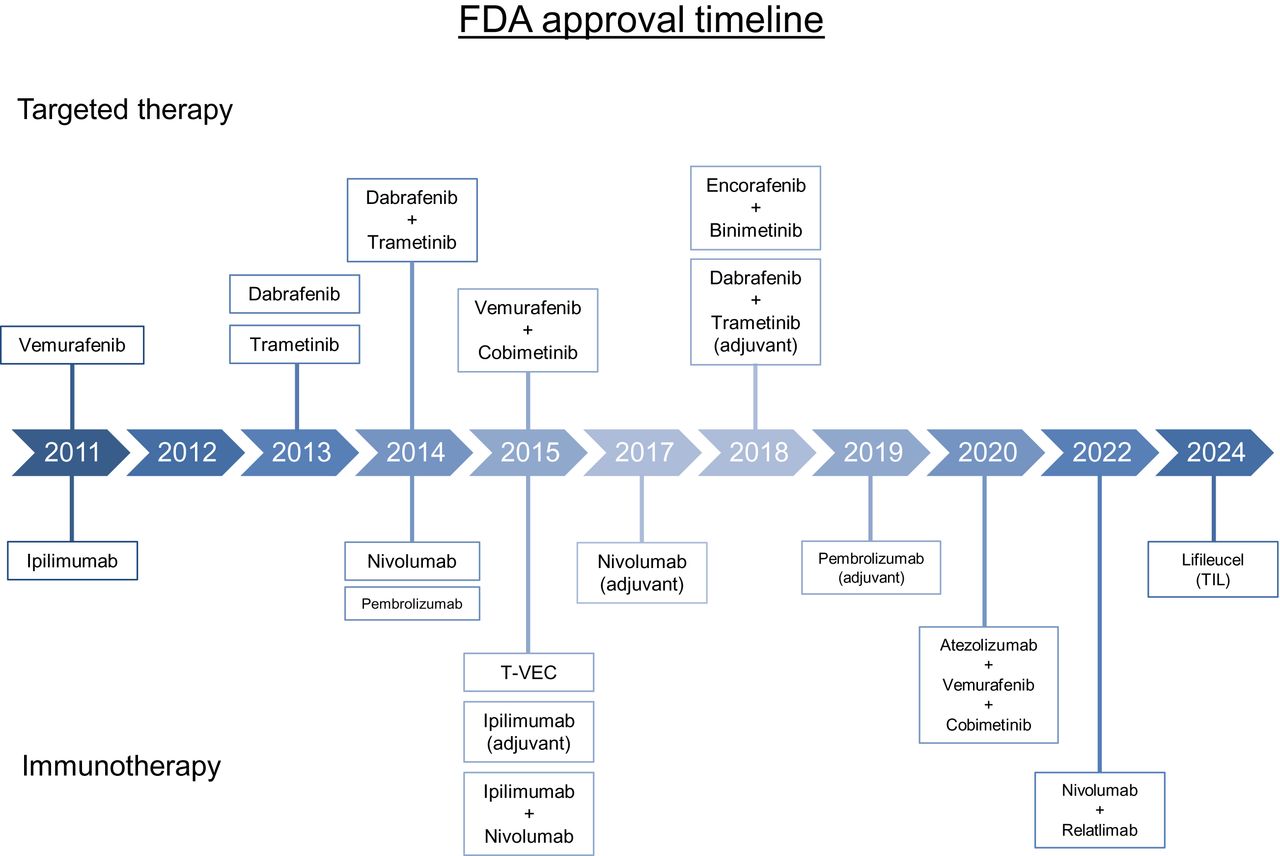
Timeline displaying FDA-approved targeted therapies and immunotherapies for melanoma. T-VEC = talimogene laherparepvec.
Before the Food and Drug Administration (FDA) approval of the BRAF inhibitor vemurafenib in 2011, limited treatment options were available for metastatic melanoma, and mortality rates were high. The efficacy of oral BRAF inhibition was first shown by Flaherty et al. when multiple patients with BRAF V600E-mutated metastatic melanoma had complete or partial tumor regression with a BRAF inhibitor (34). The role of MEK inhibition in BRAF-mutant melanoma was then established when Flaherty et al. showed that trametinib, an oral MEK inhibitor, significantly improved progression-free survival (PFS) and OS compared with chemotherapy (35). Combination BRAF and MEK inhibition with dabrafenib plus trametinib was later shown to improve PFS and OS with a reduced toxicity profile and compared with dabrafenib alone (36). Multiple regimens using combination BRAF and MEK inhibition have been approved in the interim, and they remain important options for BRAF-mutant metastatic melanoma.
After the observed success of BRAF and MEK inhibition in the metastatic setting, the role of targeted therapy was examined in the adjuvant setting. In the COMBI-AD phase III trial, 870 patients with completely resected, stage III BRAF-mutated melanoma were randomized to receive oral dabrafenib plus trametinib or placebo for 12 mo after surgery. The 5-y analysis showed that RFS and distant metastasis-free survival were both longer in patients receiving dabrafenib plus trametinib, leading to FDA approval in 2018 (37). Patients with BRAF-mutated melanoma are also eligible for adjuvant immunotherapy; the optimal adjuvant therapy has not yet been established, as combination BRAF and MEK inhibition has not been directly compared with immunotherapy. The use of neoadjuvant BRAF plus MEK inhibition remains an ongoing area of investigation.
Therapies targeting mutations other than BRAF have been shown to be efficacious for metastatic melanoma in the second-line setting (Table 1). Approximately 15%–20% of cases of acral and mucosal melanoma will harbor an activating mutation in KIT. The tyrosine kinase inhibitor imatinib has been shown to increase PFS, with an overall disease control rate of approximately 55% when studied in small clinical trials and remains an option for patients who progress while on or are ineligible for immunotherapy and harbor a KIT mutation (38). Rarely, patients with cutaneous melanoma may harbor TRK-gene fusions and can receive subsequent-line therapy with TRK-gene fusion inhibitors such as larotrectinib and entrectinib (39,40). Mutations in NRAS, an oncogene found in the MAP kinase pathway, are found in approximately 15%–20% of BRAF wild-type cutaneous melanoma. Patients harboring NRAS-mutant tumors should receive front-line immunotherapy; however, if they have evidence of disease progression, off-label use of the MEK inhibitor binimetinib can be considered in addition to clinical trial enrollment (41).
Actionable Genomic Mutations in Melanoma
IMMUNOTHERAPY AND IMMUNE CHECKPOINT INHIBITION
Advances in cancer immunotherapy have revolutionized the management of multiple malignancies, including melanoma. Studies of various forms of immunotherapy have been occurring for decades; however, it is only the recent progress in knowledge of cancer immunology and the tumor immune microenvironment that has propelled paradigm-changing treatments forward (42). Immune checkpoint inhibitors (ICIs) targeting the cytotoxic T-lymphocyte antigen-4 (CTLA-4), programmed death-1 (PD-1), and lymphocyte activation gene-3 (LAG-3) molecules are among the most prominent examples of such headway.
CTLA-4 and PD-1 are 2 key receptors expressed on T cells involved in regulating T-cell activity. CTLA-4 affects T cells during the initial stages of activation via its interaction with antigen-presenting cells. CTLA-4 competes with the T-cell costimulatory receptor cluster differentiation 28 for the binding of cluster differentiation 80 and 86, in turn decreasing T-cell activation (43). Shortly after their elucidation of the role of CTLA-4 in 1995, Allison et al. showed that blockade of the CTLA-4 molecule in mouse models resulted in enhanced antitumor immunity, spurring on multiple ensuing clinical trials in the years that would follow (44,45). PD-1 is an inhibitory receptor upregulated on activated T cells after long-term antigen exposure as part of homeostatic immune cell regulation (46). The binding of the programmed death ligand-1 (PD-L1) and programmed death ligand-2 molecules to the PD-1 receptor, which occurs primarily in chronically inflamed tissues, acts as a checkpoint for the adaptive immune system (47,48). However, tumor cells also exploit the PD-1/PD-L1 axis as a means of immune evasion. Recently, LAG-3 has emerged as a third promising target for ICIs. LAG-3 is expressed predominantly on exhausted T cells and negatively regulates T-cell activation and function. A multitude of randomized, placebo-controlled clinical trials examining the use of ICIs for the treatment of melanoma in the metastatic, adjuvant, and neoadjuvant settings has been performed after the discoveries of CTLA-4 and PD-1 by Allison and Nishimura (44,46).
IMMUNE CHECKPOINT INHIBITION IN METASTATIC MELANOMA
The FDA approved the anti–CTLA-4 monoclonal antibody ipilimumab for the treatment of metastatic melanoma in 2011 after significant improvements in OS in multiple landmark trials (Table 2) (49,50). The role of combination immunotherapy in the metastatic setting was then examined in the CheckMate 067 trial when Wolchok et al. showed that the combination of ipilimumab plus nivolumab, an anti–PD-1 monoclonal antibody, was both safe and effective (51). Data later published showed that OS was significantly increased in patients receiving ipilimumab plus nivolumab compared with ipilimumab alone (52).
ICIs for Treatment of Melanoma
The role of LAG-3 inhibition as a component of combination immunotherapy for metastatic melanoma was recently established in the RELATIVITY-047 trial (53). In patients with unresectable, stage III or IV melanoma, participants were randomly assigned to receive either relatlimab plus nivolumab or nivolumab monotherapy, both administered every 4 wk. The median PFS was 10.1 mo in the relatlimab-plus-nivolumab group compared with 4.6 mo in the nivolumab monotherapy group. OS data remain immature at this time.
ADJUVANT IMMUNE CHECKPOINT INHIBITION
The role for ICIs in the adjuvant setting was first assessed by Eggermont et al. in the EORTC 18071 trial (54). In this phase III trial of patients with completely resected, stage III disease at high risk for recurrence, administration of ipilimumab every 3 wk for 4 doses and then every 3 mo for up to 3 y after surgery resulted in significantly longer RFS, with a median RFS of 26.1 mo in the ipilimumab group versus 17.1 mo in the placebo group (hazard ratio of 0.75). Results from this trial resulted in FDA approval of ipilimumab in the adjuvant setting in 2015.
The role of PD-1 blockade in the adjuvant setting was examined in the EORTC 1325/KEYNOTE-054 trial (55). In patients with completely resected stage III melanoma, pembrolizumab, an anti-PD-1 monoclonal antibody, administered every 3 wk for a total of 18 doses, was compared with placebo administered in a similar fashion. The 18-mo RFS was 71.4% in the pembrolizumab group versus 53.2% in the placebo group; in subgroup analysis, the risk of recurrence or death was shown to be 46% lower in patients with PD-L1–positive tumors treated with pembrolizumab than in those treated with placebo. Data from updated analyses published in 2021 confirmed improvement in RFS and distant metastasis-free survival with pembrolizumab, though OS data have not yet been reported (56).
The CheckMate 238 trial compared nivolumab with ipilimumab in a head-to-head fashion; 906 patients with completely resected stage III or IV melanoma were randomized to receive either adjuvant nivolumab or ipilimumab (57). Therapy was administered for up to 1 y or until recurrence of disease or unacceptable treatment-related toxicity. Initially, results favored nivolumab, as administration resulted in significantly longer RFS and distant metastasis-free survival than for ipilimumab. However, in an updated analysis published 4 y later, there was no significant difference in OS between the 2 cohorts, although RFS remained higher in the nivolumab arm (51.7% vs. 42.5%, hazard ratio of 0.71) (58).
After significant improvements in PFS and OS with use of combination immunotherapy in the metastatic setting (59), the role of ipilimumab plus nivolumab in the adjuvant space was studied in the CheckMate 915 trial. Patients with completely resected, stage III or IV disease were randomized to receive either ipilimumab plus nivolumab or nivolumab alone for up to 1 y (60). Ipilimumab plus nivolumab did not result in an improvement in RFS compared with nivolumab monotherapy (64.6% vs. 63.2%, hazard ratio of 0.92). Furthermore, rates of grade 3 or 4 treatment-related adverse events were higher in the combination arm than in the nivolumab monotherapy arm (32.6% vs. 12.8%). The role of combination immunotherapy incorporating a LAG-3 inhibitor is currently being studied in the RELATIVITY-098 trial, in which the anti-LAG-3 agent relatlimab plus nivolumab is being compared with nivolumab monotherapy (NCT05002569).
The role of personalized medicine in adjuvant immunotherapy for melanoma is expanding. In KEYNOTE-942, Weber et al. recently showed that creation and administration of an individualized messenger RNA vaccine plus pembrolizumab improved RFS compared with pembrolizumab alone (61). The messenger RNA-4157 vaccine, generated by harvesting patient tumor samples, performing whole-exome sequencing, and identifying tumor-specific mutations, encodes up to 34 neoantigens in a lipid nanoparticle formulation. Similarly, administration of autologous tumor-infiltrating lymphocytes, initially extracted from a patient’s melanoma and then expanded ex vivo, was recently FDA-approved for metastatic melanoma previously treated with at least one line of systemic therapy (62).
NEOADJUVANT IMMUNE CHECKPOINT INHIBITION
Recently, practice-changing clinical trials exploring the role of neoadjuvant immunotherapy have demonstrated the benefit of ICIs in this setting. In the landmark phase II SWOG S1801 trial, 313 patients with resectable stage IIIB–IV melanoma with clinically detected or radiographically evident LN involvement were randomized to receive pembrolizumab, administered every 3 wk for a total of 3 doses before surgery, followed by 15 doses of pembrolizumab as adjuvant therapy or surgical resection followed by adjuvant pembrolizumab every 3 wk for a total of 18 doses (63). At the median follow-up of 15 mo, patients who received neoadjuvant pembrolizumab had improved EFS compared with those who received adjuvant therapy alone, with 2-y EFS of 72% versus 49%, respectively, with a hazard ratio of 0.58. The benefit of neoadjuvant pembrolizumab was seen across all subgroups, including patients with BRAF-mutated and wild-type disease, and treatment-related toxicities resulting in an inability to undergo surgery occurred in less than 10% of patients. OS data from this trial remain immature at this time.
IMAGING SURVEILLANCE OF CUTANEOUS MELANOMA
Given the historical lack of effective treatments for melanoma, there has been a paucity of randomized clinical trials conducted on imaging surveillance. As such, little evidence currently exists regarding optimal modality or timeline. As a result, the role of imaging surveillance in melanoma lacks expert agreement, reflected in national and international guideline heterogeneity. Consistent among guidelines, however, is the recommendation against surveillance imaging in early-stage disease, for which imaging detection rates have been shown to be lower than false-positive rates (64). Most guidelines do recommend surveillance imaging with stage IIB or IIC disease, for which rates of distant recurrence are significantly increased (18).
Regarding preferred imaging modality, evidence for the improved accuracy of PET/CT compared with conventional contrast-enhanced CT in melanoma surveillance is strong. A 2011 systematic review demonstrated that the sensitivity and specificity of PET/CT was 86% and 91%, compared with 63% and 78% for CT of the chest, abdomen, and pelvis (65). The continued recommendation for CT of the chest, abdomen, and pelvis by some guidelines has been explained as a reflection of the limited availability of PET/CT in certain regions and demographics. Contrast-enhanced CT, and potentially whole-body MRI, can be considered in this context.
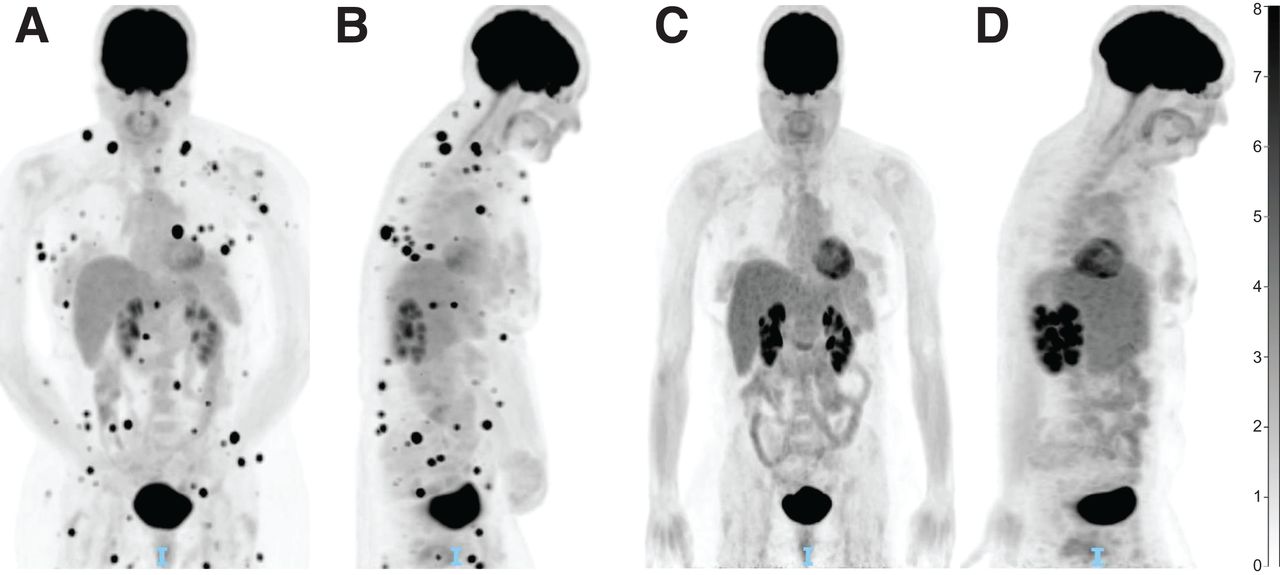
In patients treated with ICI, 18F-FDG PET appears to be particularly useful in the monitoring and prediction of response. Using RECIST, the most validated CT criteria in use for solid tumors, multiple 18F-FDG PET criteria have been adapted for the interpretation of immunotherapy response, most recently with PET Response Evaluation Criteria for Immunotherapy and immune PERCIST. Though validation is ongoing, all of these criteria ultimately conclude that metabolic response in 18F-FDG PET is strongly associated with survival outcomes (66). The recently published joint guidelines from the European Association of Nuclear Medicine, Society of Nuclear Medicine and Molecular Imaging, and Australian and New Zealand Society of Nuclear Medicine therefore endorse 18F-FDG PET/CT as the imaging modality of choice for baseline disease assessment before the start of immunotherapy. 18F-FDG PET/CT is also recommended after approximately 3–4 cycles of treatment (Fig. 3) in the case of clinical deterioration or suspected progression on other imaging, before restarting treatment in cases of temporary interruption, and before treatment discontinuation (67).

PET/CT for immunotherapy treatment response assessment. (A) PET/CT displaying innumerable intramuscular and subcutaneous soft-tissue nodules consistent with metastatic melanoma (coronal view). (B) Initial PET/CT (midsagittal view). (C) Repeat PET/CT less than 3 mo later and after 3 cycles of ipilimumab-nivolumab, showing complete resolution of soft-tissue nodules (coronal view). (D) Repeat PET/CT displaying treatment response (midsagittal view). Unit of measure for intensity bar is SUV = C(T)/[injection dose (MBq)/patient weight (kg)]).
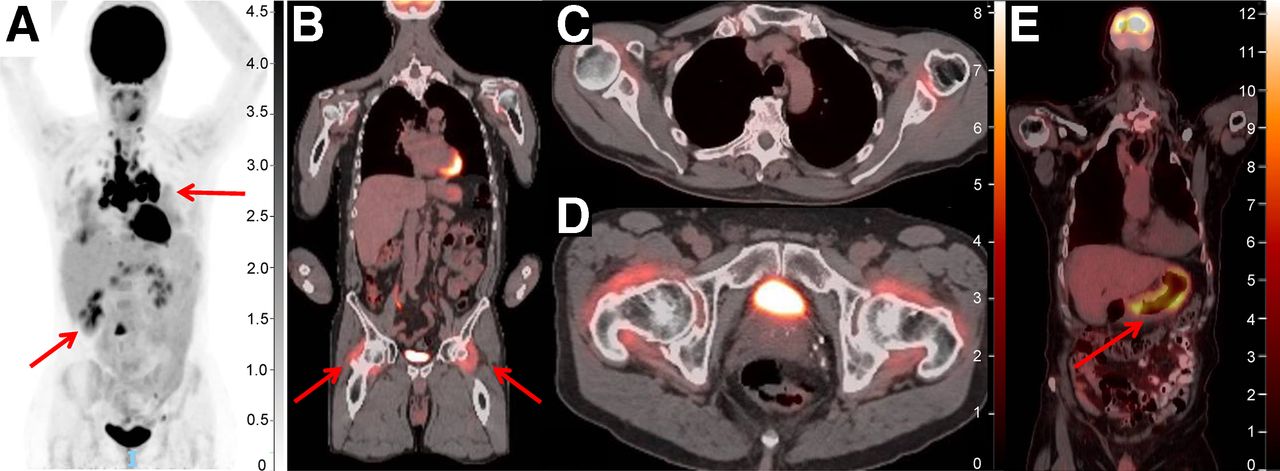
Given the novel mechanisms of action of ICIs, atypical treatment responses not seen with use of traditional chemotherapy or other targeted therapies have been observed; these include pseudoprogression, hyperprogression, and immune-related adverse events. 18F-FDG PET/CT is again uniquely advantageous in this context given its ability to characterize metabolically active tissue. Pseudoprogression, defined as an initial transient increase in tumor metabolic activity and morphologic size followed by a true response, occurs in approximately 10% of patients treated with ICIs. It is seen mostly frequently within the first 4–6 wk after initiation of treatment but can occur up to several months thereafter (67). Differentiating pseudoprogression from true progression requires follow-up imaging 4–8 wk later. Conversely, hyperprogression—an atypical, true acceleration of tumor growth on imaging after initiation of immunotherapy—represents rapid treatment failure and has been observed in melanoma and other malignancies. Efforts to define and understand the mechanism of this phenomenon continue; however, it is thought to occur in approximately 4%–26% of cases when defined by doubling of volume or growth rate on imaging (68,69). Finally, 18F-FDG PET has also been shown to be uniquely effective in identifying immune-related adverse events, including detection before onset of symptoms (Fig. 4) (67).

PET/CT for detection of immune-related adverse events. (A) PET/CT displaying hypermetabolic pulmonary nodules, subcarinal and hilar adenopathy, and hypermetabolic liver lesions (arrows), initially concerning for metastatic disease but later biopsied and found to be consistent with ICI-related sarcoidosis. (B) Increased 18F-FDG uptake bilaterally in hips (arrows) and shoulders consistent with immune-mediated arthritis. (C and D) Two axial views of 18F-FDG uptake in shoulders and hips. (E) Diffuse 18F-FDG uptake within posterior and posterior-lateral gastric wall consistent with immune-mediated gastritis, noted before onset of symptoms. Unit of measure for intensity bar is SUV = C(T)/[injection dose (MBq)/patient weight (kg)]).
FUTURE OF RADIOLOGY IN MELANOMA
Recently, the use of high-intensity focused ultrasound as a noninvasive form of tumor ablation has been proposed as an adjunct therapy for multiple solid tumors, including melanoma. An ultrasound transducer positioned outside the body or within a body cavity is used to focus high-intensity ultrasound beams in a small region of tumor, resulting in extreme intratumoral temperature elevations in a matter of seconds, ultimately causing necrosis of tumor cells (70). In addition to a reduction in tumor burden, high-intensity focused ultrasound results in the release of tumor antigens and damage-associated molecular patterns, triggering both innate and adaptive immune responses. Studies evaluating optimization of high-intensity focused ultrasound protocols and incorporation of immunotherapy in addition to high-intensity focused ultrasound are ongoing (NCT04116320).
Current investigations into the role of artificial intelligence and machine learning in 18F-FDG PET/CT are promising. One area in which machine learning has proven particularly useful is in the identification of novel metabolic markers on 18F-FDG PET to guide treatment strategies. Multiple studies on one such parameter, total metabolic tumor volume, calculated by both manual and automated lesion segmentation, have demonstrated significant correlation with poor treatment response to pembrolizumab (71). Another marker with apparent significant prognostic value is high metabolic activity of hematopoietic tissues, such as bone marrow and spleen, which has been correlated with poor response to ICIs and an immunosuppressive environment in multiple studies (72).
The development of novel radiolabeled tracers to detect malignancy, guide therapy, and identify cellular microenvironments is ongoing. Numerous tracers are being studied in various preclinical and clinical stages of development. Melanin imaging, although suggested in recent studies to be a more complex target than initially thought, is highly specific to melanoma patients. The labeling of 18F to benzamide derivatives such as 5-FPN (18F-5-fluoro-N-[2-(diethylamino)ethyl]picolinamide) and MEL050 (18F-6-fluoro-N-[2-(diethylamino)ethyl]pyridine-3-carboxamide) has demonstrated in vivo imaging performance superior to 18F-FDG (73). Melanin-targeted therapy has also demonstrated early promise, with 131I and 188Re labeled to various molecules demonstrating antitumor efficacy with limited toxicities (73). Multiple tracers, including 68Ga, 89Zr, and 18F, have also been attached to monoclonal antibodies against PD-1 and PD-L1, allowing for a noninvasive whole-body map of immune checkpoint proteins. These radiolabeled tracers have the potential to be used to stratify treatment candidates, monitor therapy, and create a pathway for targeted therapy (74).
Footnotes
Learning Objectives: On successful completion of this activity, participants should be able to (1) describe the current guidelines for baseline imaging studies for patients with newly diagnosed melanoma; (2) identify the biologic targets of the Food and Drug Administration–approved checkpoint inhibitors used in the treatment of advanced melanoma; and (3) consider the optimal use of PET/CT in surveillance of treated melanoma.
Financial Disclosure: Dr. Gaughan’s institution has received research funding from Merck, Bristol Myers Squibb, Iovance, and Regeneron. Dr. Gaughan received travel reimbursement from Iovance and Regeneron. This work was supported by UVA Cancer support grant P30CA044579. The authors of this article have indicated no other relevant relationships that could be perceived as a real or apparent conflict of interest.
CME Credit: SNMMI is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to sponsor continuing education for physicians. SNMMI designates each JNM continuing education article for a maximum of 2.0 AMA PRA Category 1 Credits. Physicians should claim only credit commensurate with the extent of their participation in the activity. For CE credit, SAM, and other credit types, participants can access this activity through the SNMMI website (http://www.snmmilearningcenter.org) through December 2027.
Published online Nov. 14, 2024.
- © 2024 by the Society of Nuclear Medicine and Molecular Imaging.
Immediate Open Access: Creative Commons Attribution 4.0 International License (CC BY) allows users to share and adapt with attribution, excluding materials credited to previous publications. License: https://creativecommons.org/licenses/by/4.0/. Details: https://jnm.snmjournals.org/page/permissions.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.
- 76.
- 77.
- 78.
- 79.
- 80.
- Received for publication March 25, 2024.
- Accepted for publication October 22, 2024.
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- EPIDEMIOLOGY
- STAGING OF DISEASE AT TIME OF DIAGNOSIS
- MANAGEMENT OF PRIMARY CUTANEOUS MELANOMA
- MOLECULAR PROFILING AND TARGETED THERAPIES
- IMMUNOTHERAPY AND IMMUNE CHECKPOINT INHIBITION
- IMMUNE CHECKPOINT INHIBITION IN METASTATIC MELANOMA
- ADJUVANT IMMUNE CHECKPOINT INHIBITION
- NEOADJUVANT IMMUNE CHECKPOINT INHIBITION
- IMAGING SURVEILLANCE OF CUTANEOUS MELANOMA
- FUTURE OF RADIOLOGY IN MELANOMA
- Footnotes
- REFERENCES
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.