


Abstract
Copper is an element required for cell proliferation and angiogenesis. Human prostate cancer xenografts with increased 64Cu radioactivity were visualized previously by PET using 64CuCl2 as a radiotracer (64CuCl2 PET). This study aimed to determine whether the increased tumor 64Cu radioactivity was due to increased cellular uptake of 64Cu mediated by human copper transporter 1 (hCtr1) or simply due to nonspecific binding of ionic 64CuCl2 to tumor tissue. In addition, the functional role of hCtr1 in proliferation of prostate cancer cells and tumor growth was also assessed. Methods: A lentiviral vector encoding short-hairpin RNA specific for hCtr1 (Lenti-hCtr1-shRNA) was constructed for RNA interference–mediated knockdown of hCtr1 expression in prostate cancer cells. The degree of hCtr1 knockdown was determined by Western blot, and the effect of hCtr1 knockdown on copper uptake and proliferation were examined in vitro by cellular 64Cu uptake and cell proliferation assays. The effects of hCtr1 knockdown on tumor uptake of 64Cu were determined by PET quantification and tissue radioactivity assay. The effects of hCtr1 knockdown on tumor growth were assessed by PET/CT and tumor size measurement with a caliper. Results: RNA interference–mediated knockdown of hCtr1 was associated with the reduced cellular uptake of 64Cu and the suppression of prostate cancer cell proliferation in vitro. At 24 h after intravenous injection of the tracer 64CuCl2, the 64Cu uptake by the tumors with knockdown of hCtr1 (4.02 ± 0.31 percentage injected dose per gram [%ID/g] in Lenti-hCtr1-shRNA-PC-3 and 2.30 ± 0.59 %ID/g in Lenti-hCtr1-shRNA-DU-145) was significantly lower than the 64Cu uptake by the control tumors without knockdown of hCtr1 (7.21 ± 1.48 %ID/g in Lenti-SCR-shRNA-PC-3 and 5.57 ± 1.20 %ID/g in Lenti-SCR-shRNA-DU-145, P < 0.001) by PET quantification. Moreover, the volumes of prostate cancer xenograft tumors with knockdown of hCtr1 (179 ± 111 mm3 for Lenti-hCtr1-shRNA-PC-3 or 39 ± 22 mm3 for Lenti-hCtr1-shRNA-DU-145) were significantly smaller than those without knockdown of hCtr1 (536 ± 191 mm3 for Lenti- SCR-shRNA-PC-3 or 208 ± 104 mm3 for Lenti-SCR-shRNA-DU-145, P < 0.01). Conclusion: Overall, data indicated that hCtr1 is a promising theranostic target, which can be further developed for metabolic imaging of prostate cancer using 64CuCl2 PET/CT and personalized cancer therapy targeting copper metabolism.
Prostate cancer is a complex, genetically heterogeneous disease with variable clinical manifestations (1,2). PET is useful for the noninvasive assessment of metabolic activities of prostate cancer and monitoring response to treatment (3). Multiple radiopharmaceuticals were developed for PET of various metabolic pathways in prostate cancer, such as metabolism of glucose, lipid, and choline and synthesis of DNA and protein molecules. These radiotracers include 18F-FDG (4), 11C-choline (5), 18F-choline (6), 18F-fluoroethylcholine (7), 11C-acetate (8), 18F-FMAU (FMAU is 1-(2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)thymine) (9), and 11C-methionine (10). Given the genetic and metabolic heterogeneity of prostate cancer, there is need for additional radiotracers for PET imaging of other metabolic pathways in prostate cancer.
Copper is known to be critical for cell proliferation, angiogenesis, and tumor growth (11–13), and increased copper ions were detected in cancer tissues (14,15). Exploring copper metabolism as an imaging biomarker, we demonstrated that human prostate cancer xenografts with increased 64Cu radioactivity could be visualized by PET using 64CuCl2 as a tracer (16). The expression of human copper transporter 1 (hCtr1), a high-affinity influx copper transporter (17), was demonstrated in prostate cancer xenograft tissues by immunohistochemistry analysis (16). Using RNA interference (RNAi) technology (18), this study aimed to determine whether increased 64Cu radioactivity in prostate cancer xenografts visualized by PET was caused by increased cellular uptake of 64Cu mediated by influx copper transporter activity of hCtr1. The effect of hCtr1 knockdown on cell proliferation was examined in vitro, and the effect of hCtr1 knockdown on tumor growth was examined in vivo by comparing the size of the tumors with knockdown of hCtr1 with the size of the tumors without knockdown of hCtr1. Overall, data support hCtr1 as a new biomarker for metabolic imaging of human prostate cancer using 64CuCl2 PET/CT and as a promising target for prostate cancer therapy targeting copper metabolism.
MATERIALS AND METHODS
Cells, Reagents, and Radiopharmaceuticals
The human prostate cancer cell line PC-3 (androgen receptor–negative) and C4-2 (androgen receptor–positive), a subline of LNCaP cells (American Type Culture Collection [ATCC]), were cultured in T-medium (Life Technologies) supplemented with 5% fetal bovine serum (FBS) and antibiotics (penicillin [100 U/mL] and streptomycin [100 mg/mL]) from Biosource International. The human prostate cancer cell line Du-145 (androgen receptor–negative), purchased from ATCC, was cultured in RPMI1640 medium supplemented with 10% FBS and antibiotics. Normal prostate epithelia cells (Lonza) and immortalized prostate epithelial cells (RWPE-1, RWPE-2, and PZ-HPV-7), purchased from ATCC, were cultured in prostate epithelial cell growth medium (PrEGM) supplemented with growth factors, cytokines, and supplements supplied in the SingleQuots kit (Lonza), 10% FBS, and antibiotics. Recombinant hCtr1 protein (19) was a gift from Vinzenz M. Unger at Yale University. Polyclonal antibody specific for hCtr1 was prepared using a recombinant hCtr1 protein derived from the first 67 amino acid residues at the N terminus of hCtr1 (17,20), as described in the supplemental material (available at http://jnm.snmjournals.org). The 64Cu radionuclide (half-life, 12.7 h; decay characteristics β+, 19%, and β−, 40%) was produced in a cyclotron with a radionuclide purity greater than 99% and was supplied as 64CuCl2 in an HCl solution (0.1 mol/L) by the Mallinckrodt Institute of Radiology, Washington University.
Construction of hCtr1 Short Hairpin RNA (shRNA) Lentiviral Vector
A lentiviral vector encoding hCtr1 shRNA (Lenti-hCtr1-shRNA virus) was constructed as described previously (21,22). Briefly, 4 plasmid vectors encoding shRNA specific for hCtr1 and 1 plasmid encoding nonspecific, scrambled shRNA were constructed as described in the supplemental material. The hCtr1 shRNA sequence (GGAGTACACTTTCATGTGATT) was polymerase chain reaction (PCR)–amplified from the hCtr1 shRNA plasmid vector 2 and inserted into the BamHI and EcoRI site of a pGreenPurolenti-shRNA expression vector (System Biosciences). This lentiviral vector contains a puromycin-resistance gene to enable drug selection of target cells stably expressing the siRNA after transduction of the cells with lentivirus-encoding shRNA. At 72 h after transfection of 293 cells with a mixture of pLenti-hCtr1-shRNA and lentiviral packaging plasmid DNA, the supernatants containing lentivirus particles were harvested and the titers (pfu/mL) of Lenti-hCtr1-shRNA virus were determined, using an Ultra Rapid lentiviral titer kit (System Biosciences). Stock solution of the Lenti-hCtr1-shRNA virus was stored at −80°C for use within 6 months after preparation. A negative control lentiviral vector (Lenti-SCR-shRNA virus) encoding scrambled (SCR) shRNA sequence (Supplemental Table 1) was also prepared using the same method as described above. The expression of hCtr1 in the cells transduced by the lentivirus was examined by Western blot in a method as described in the supplemental material.
Cell 64Cu Uptake and Proliferation Assays
To assess cellular copper uptake mediated by hCtr1, the cells were seeded into a 24-well plate (5 × 104 cells/well) and cultured in medium without serum under a serum starvation condition for 24 h. After incubation of the cells with 64CuCl2 (74 kBq or 2 μCi/well) at 37°C for 1 h, the cells were harvested and washed 3 times with cold phosphate-buffered saline. The radioactivity of the cells was counted with a Packard Cobra II Gamma Counter (Perkin-Elmer). A cell proliferation assay was conducted using a CCK-8 Cell Proliferation Assay kit (Dojindo) as described in the supplemental material, based on the method described previously (23). The experiment was conducted in triplicate for each point and repeated 3 times.
Small-Animal PET/CT
All animal experiments were conducted under the protocol approved by the University of Texas Southwestern Institutional Animal Care and Use Committee. PET of athymic nu/nu mice (male; age, 4–5 wks) bearing human prostate cancer xenografts was performed using a Siemens Inveon PET/CT Multimodality System as described previously (16,24). Briefly, a structural CT scan of tumor-bearing mice was acquired (80 kV, 500 μA) with a pixel size of approximately 0.1 mm to create an anatomic image that was subsequently used for attenuation correction of the PET emission data. After conclusion of the CT scan, mice were injected with the tracer 64CuCl2 (74 kBq or 2 μCi/g of body weight) intravenously via the tail vein. Static whole-body imaging was performed at 2 and 24 h after intravenous injection of the tracer, which consisted of 2 overlapping frames of 15 min for each frame. On completion of the PET/CT at 24 h after injection, a tissue radioactivity assay was performed, and tissue radioactivity was calculated and expressed as decay-corrected percentage injected dose per gram of tissue (%ID/g) as described previously (16). The size of the postmortem tumors was measured with a caliper, and tumor volumes were calculated using an ellipsoidal formula (1/2 × (length × width2)) modified from that described previously (25).
PET Quantitative Analysis
PET images were reconstructed using the ordered-subsets expectation maximization 3-dimensional algorithm and analyzed using the Inveon Research Workplace (IRW) software (Siemens), which allows fusion of CT and PET image volumes, the reslicing of fused images into arbitrary views, and the definition of regions of interest. Static whole-body images obtained at 2 and 24 h were converted to decay-corrected images representing the %ID/g by normalizing the activity concentration in each pixel (MBq/cm3) by the injected activity (MBq) and multiplying the result by 100%. Moreover, we used the conversion 1 cm3 = 1 g.
Statistical Analysis
Independent sample t tests were applied to assess significant differences in cellular 64Cu uptake and cell proliferation in vitro between the cells with or without knockdown of hCtr1. Moreover, paired t tests were applied to assess significant differences in tumor 64Cu uptake (ID%/g) and volume between prostate cancer xenografts with or without knockdown of hCtr1. A P value of less than 0.05 was considered to represent statistical significance.
RESULTS
Expression of hCtr1 in Prostate Cancer Cells
Polyclonal antibodies specific for hCtr1 were obtained by immunization of rabbits with recombinant hCtr1 protein encompassing the first 67 amino acid residues of extracellular domain of hCtr1 and affinity chromatography purification of hCtr1 antibodies from rabbit antiserum (Fig. 1A). The expression of hCtr1 in prostate cancer cells (PC-3, Du-145, and C4-2), immortalized prostate epithelial cells containing HPV oncogene (RWPE-1, RWPE-2, and PZ-HPV-7), and normal prostate epithelial cells was detected by Western blot using polyclonal antibodies specific for hCtr1 (Fig. 1B). Densitometry semiquantitative analysis of Western blot demonstrated elevated expression of hCtr1 in prostate cancer cells (327% higher in PC-3, 425% higher in DU-145, 408% higher in C4-2) and immortalized prostate epithelial cells (279% higher in PZ-HPV-7, 386% higher in RWPE-1, and 349% higher in RWPE-2), compared with expression of hCtr1 (set as 100%) in normal prostate epithelial cells.

Expression of hCtr1 in prostate cancer cells by Western Blot. (A) Schematic presentation of amino acid sequence of recombinant hCtr1 protein used for preparation of polyclonal antibody specific for hCtr1. (B) Western blot of hCtr1 in prostate cancer cells (lane 1, PC-3; lane 2, DU-145; lane 3, C4-2), immortalized prostate epithelial cells containing HPV oncogene (lane 4, PZ-HPV-7; lane 5, RWPE-1; lane 6, RWPE-2), and normal prostate epithelial cells (lane 7). Loading amount was 60 μg of total cellular protein/lane for lanes 1–7. Rec hCtr1 = recombinant hCtr1 protein (30 ng/lane).
Reduction of Copper Uptake and Suppression of Cell Proliferation by Knockdown of hCtr1
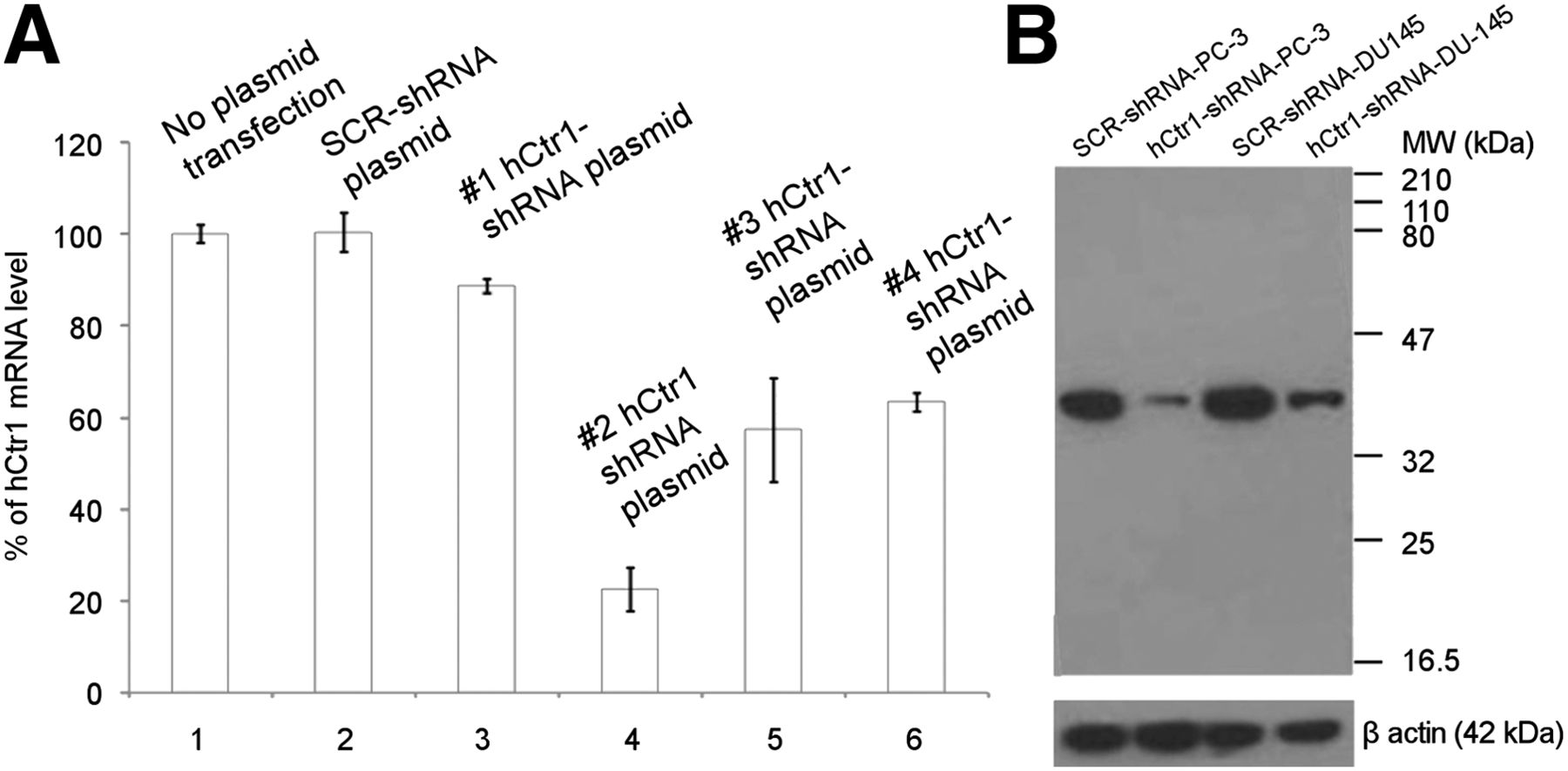
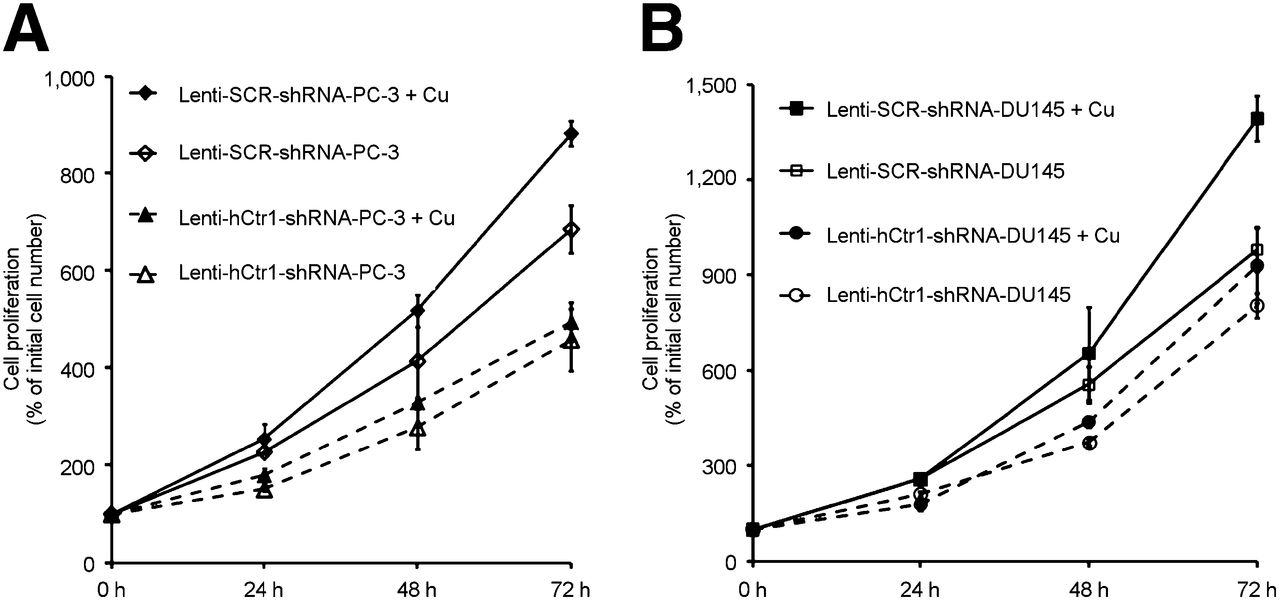
Four plasmid vectors encoding shRNA sequence targeting different regions of the hCtr1 gene were constructed for knockdown of hCtr1 expression in prostate cancer cells. As shown in Figure 2A, the #2 hCtr1 shRNA plasmid showed highest efficacy for knocking down the endogenous hCtr1 in prostate cancer cells, among 4 different hCtr1 shRNA plasmid vectors tested. hCtr1 messenger RNA (mRNA) level in the puromycin-resistant PC-3 cells transfected with the #2 hCtr1 shRNA plasmid was determined to be 22% of hCtr1 mRNA detected in the puromycin-resistant PC-3 cells transfected with SCR-shRNA plasmid or PC-3 cells without plasmid transfection by a real-time quantitative reverse transcription-PCR assay (qRT-PCR). The level of hCtr1 mRNA detected in the puromycin-resistant PC-3 cells transfected with #2 hCtr1 shRNA plasmid was determined to be 22% of the hCtr1 mRNA level detected in the puromycin-resistant PC-3 cells transfected with SCR-shRNA plasmid or PC-3 cells without plasmid transfection by qRT-PCR. Meanwhile, the levels of hCtr1 mRNA level detected in the puromycin-resistant PC-3 cells transfected with the other 3 hCtr1 shRNA plasmids (#1, #3, and #4 hCtr1 shRNA plasmids) were determined to be 89%, 57%, and 63% of the hCtr1 mRNA level detected in the puromycin-resistant PC-3 cells transfected with SCR-shRNA plasmid or PC-3 cells without plasmid transfection by qRT-PCR, respectively. A Lenti-viral vector containing hCtr1 shRNA sequence (GGAGTACACTTTCATGTGATT) derived from #2 hCtr1 shRNA plasmid, Lenti-hCtr1-shRNA virus, was constructed and used for constant knockdown of hCtr1 in prostate cancer cells. A Lenti-viral vector encoding scrambled shRNA sequence, Lenti-SCR-shRNA virus, was constructed and used as a control. The expression of hCtr1 in the puromycin-resistant PC-3 cells transduced with Lenti-hCtr1-shRNA virus (Lenti-hCtr1-shRNA-PC-3 cells) was knockdown to 20% of hCtr1 expression in the puromycin-resistant PC-3 cells transduced with Lenti-SCR-shRNA virus (Lenti-SCR-shRNA-PC-3 cells) by Western blot (Fig. 2B). Similarly, the expression of hCtr1 in the puromycin-resistant DU-145 cells transduced with Lenti-hCtr1-shRNA virus (Lenti-hCtr1-shRNA-DU145 cells) was knockdown to 25% of hCtr1 expression in the puromycin-resistant DU145 cells transduced with Lenti-SCR-shRNA virus (Lenti-SCR-shRNA-DU145 cells, Fig. 2B). Knockdown of hCtr1 resulted in the reduction of copper uptake by these cells (Fig. 3). After incubation with 64CuCl2 (74 kBq or 2 μCi/well) for 24 h, 64Cu radioactivity of the Lenti-hCtr1-shRNA-PC-3 cells (154 ± 7 cpm/104 cells) or Lenti-hCtr1-shRNA-DU-145 cells (108 ± 17 cpm/104 cells) was lower than 64Cu radioactivity of the control Lenti-SCR-shRNA-PC-3 cells (585 ± 52 cpm/104 cells, P = 0.04) or Lenti-SCR-shRNA-DU-145 cells (481 ± 23 cpm/104 cells, P < 0.01). Furthermore, knockdown of hCtr1 resulted in slower proliferation of PC-3 and DU-145 cells (Fig. 4). At the end of the 72-h culture, there was a significant difference between the proliferation of the cells with knockdown of hCtr1 (458% ± 64% of inoculated cells for Lenti-hCtr1-shRNA-PC-3 cells and 804% ± 39% of inoculated cells for Lenti-hCtr1-shRNA-DU-145 cells) and the cells without knockdown of hCtr1 (686% ± 49% of inoculated cells for Lenti-SCR-shRNA-PC-3 cells, P = 0.01, and 981% ± 70% of inoculated cells for Lenti-SCR-shRNA-DU-145 cells, P = 0.03). When cultured in cell culture medium containing FBS supplemented with 10 μM CuCl2 after serum starvation, the proliferation of Lenti-SCR-shRNA-PC-3 cells (883% ± 33% of inoculated cells) or Lenti-SCR-shRNA-DU-145 cells (1,393% ± 70% of inoculated cells) was significantly enhanced (P < 0.01), compared with proliferation of Lenti-SCR-shRNA-PC-3 cells (686% ± 49%) and Lenti-SCR-shRNA-DU-145 cells (981% ± 70%) cultured in cell culture medium containing FBS without copper supplement. However, the proliferation of Lenti-hCtr1-shRNA-PC-3 cells (495% ± 24% of inoculated cells) or Lenti-hCtr1-shRNA-DU-145 cells (930% ± 118% of inoculated cells) was not changed when cultured in cell culture medium containing FBS supplemented with 10 μM CuCl2, compared with proliferation of these cells cultured in cell culture medium containing FBS without copper supplement (458% ± 64% of inoculated cells for Lenti-hCtr1-shRNA-PC-3 cells and 804% ± 39% of inoculated cells for Lenti-hCtr1-shRNA-DU-145 cells), indicating hCtr1 is critical for copper-stimulated proliferation of prostate cancer cells.

Knockdown of hCtr1 in prostate cancer cells by qRT-PCR and Western blot. (A) Quantity of hCtr1 mRNA in puromycin-resistant PC-3 cells transfected with plasmid encoding hCtr1 shRNA was determined by qRT-PCR and compared with hCtr1 mRNA in wild-type PC-3 cells without plasmid transfection and puromycin-resistant PC-3 cells transfected with SCR-shRNA plasmid. Among 4 hCtr1-shRNA plasmids, #2 hCtr1-shRNA plasmid showed highest efficacy for knocking down hCtr1 in PC-3 cells. (B) Knockdown of hCtr1 in puromycin-resistant PC-3 and DU-145 prostate cancer cells transduced with Lenti-hCtr1-shRNA virus encoding hCtr1 shRNA sequence derived from #2 hCtr1-shRNA plasmid by Western blot. SCR-shRNA PC-3, control Lenti-SCR-shRNA PC-3 cells; hCtr1-shRNA PC-3, Lenti-hCtr1-shRNA PC-3 cells; SCR-shRNA DU-145, control Lenti-SCR-shRNA DU-145 cells; hCtr1-shRNA DU-145, Lenti-hCtr1-shRNA DU-145 cells. Loading amount for lanes 1–4 = 60 μg/lane. Error bar, SD.

Reduced cellular copper uptake by prostate cancer cells after knocking down hCtr1 expression. 64Cu radioactivity of cells with knockdown of hCtr1 (Lenti-hCtr1-shRNA-PC-3 cells and Lenti-hCtr1-shRNA-DU145 cells) was significantly lower than that of cells without knockdown of hCtr1 (Lenti-SCR-shRNA-PC-3 cells and Lenti-SCR-shRNA-DU-145 cells), at 1 h after incubation with 64CuCl2. Error bar, SD.

Effects of hCtr1 knockdown on prostate cancer cell proliferation in vitro. (A) Proliferation of PC-3 cells with knockdown of hCtr1 (Lenti-hCtr1-shRNA-PC-3 cells) was significantly slower than proliferation of cells without knockdown of hCtr1 (Lenti-SCR-shRNA-PC-3 cells), when cultured in cell culture medium containing FBS with or without copper supplement. (B) Proliferation of DU-145 cells with knockdown of hCtr1 (Lenti-hCtr1-shRNA-DU145 cells) was slower than proliferation of DU-145 cells without knockdown of hCtr1 (Lenti-SCR-shRNA-DU145 cells), when cultured in cell culture medium containing FBS with or without copper supplement. Error bar, SD.
Reduced Tumor 64Cu Uptake and Growth Inhibition by Knockdown of hCtr1
Using PET/CT imaging, we determined a significant reduction of 64Cu uptake in both PC-3 and DU-145 tumors with knockdown of hCtr1 (Lenti-hCtr1-shRNA-PC-3 tumors and Lenti-hCtr1-shRNA-DU145 tumors), as compared with 64Cu uptake by the tumors without knockdown of hCtr1 (Lenti-SCR-shRNA-PC-3 tumors or Lenti-SCR-shRNA-DU-145 tumors; Figs. 5 and 6). At 2 h after injection, the 64Cu radioactivity of the Lenti-hCtr1-shRNA-PC-3 tumors was determined as (1.35 ± 0.08 %ID/g) by PET quantification, which was significantly lower than the 64Cu radioactivity of the control Lenti-SCR-shRNA-PC-3 tumors (2.10 ± 0.36 %ID/g, P = 0.02). Moreover, the 64Cu radioactivity of the Lenti-hCtr1-shRNA-DU-145 tumors was determined as 1.51 ± 0.30 %ID/g, which was also significantly lower than the 64Cu radioactivity of the control Lenti-SCR-shRNA-DU-145 tumors (4.03 ± 0.80 %ID/g, P < 0.001). The decrease in 64Cu uptake was even more obvious in PET images obtained at 24 h after injection. At 24 h after injection, the 64Cu radioactivity of the Lenti-hCtr1-shRNA-PC-3 tumors (4.02 ± 0.31 %ID/g) was significantly lower than that of the control Lenti-SCR-shRNA-PC-3 tumors (7.21 ± 1.48 %ID/g, P < 0.001), as was the case in DU-145 tumors (2.30 ± 0.59 %ID/g in Lenti-hCtr1-shRNA-DU-145 tumors vs. 5.57 ± 1.20 %ID/g in control Lenti-SCR-shRNA-DU-145 tumors; P < 0.001). The findings of PET quantitative analysis were confirmed by tumor 64Cu radioactivity from postmortem tissue radioactivity assay (Table 1). Knockdown of hCtr1 was associated with tumor growth inhibition as visualized on PET/CT (Fig. 5) and ex vivo tumor size measurement (Fig. 7A). The tumor volume of Lenti-hCtr1-shRNA PC-3 tumors (179 ± 111 mm3) was found to be significantly smaller than that of Lenti-SCR-shRNA PC-3 tumors (536 ± 191 mm3, P < 0.01) at 4 wks after implantation of the cells (Fig. 7B). Similarly, tumor volume of Lenti-hCtr1-shRNA-DU-145 tumors (39 ± 22 mm3) was found to be significantly smaller than that of Lenti-SCR-shRNA DU-145 tumors (208 ± 104 mm3, P < 0.01) at 7 wks after implantation of the cells (Fig. 7B).
Biodistribution of 64Cu in Tumor-Bearing Mice at 24 Hours After Injection of 64CuCl2

PET/CT of tumor-bearing mice injected with 64CuCl2 as tracer. (A) Reduced 64Cu radioactivity and growth inhibition of PC-3 tumor with knockdown of hCtr1 (Lenti-hCtr1-shRNA-PC-3 tumor on right, marked by *), compared with PC-3 tumors without knockdown of hCtr1 (Lenti-SCR-shRNA-PC-3 on left marked by **). (B) Reduced 64Cu radioactivity and growth inhibition of DU-145 tumor with knockdown of hCtr1 (Lenti-hCtr1-shRNA-DU-145 tumor on right marked by *), compared with DU-145 tumors without knockdown of hCtr1 (Lenti-SCR-shRNA-DU-145 tumor on left marked by **). p.i. = postintravenous injection.

Quantification of tumor 64Cu uptake by PET/CT of tumor-bearing mice injected with 64CuCl2 as tracer. (A) 64Cu radioactivity of PC-3 tumors with knockdown of hCtr1 (Lenti-hCtr1-shRNA-PC-3 tumor) was significantly lower than that of PC-3 tumors without knockdown of hCtr1 (Lenit-SCR-shRNA-PC-3 tumor). (B) 64Cu radioactivity of DU-145 tumors with knockdown of hCtr1 (Lenti-hCtr1-shRNA-DU-145 tumor) was significantly lower than that of DU-145 tumor without knockdown of hCtr1 (Lenti-SCR-shRNA-DU-145 tumor). Error bar, SD.

Suppression of tumor growth by knockdown of hCtr1 in human prostate cancer xenografts in mice. (A) Tumors with knockdown of hCtr1 (Lenti-hCtr1-shRNA-PC-3 tumor and Lenti-hCtr1-shRNA-DU-145 tumor) were smaller than those without knockdown of hCtr1 (Lenti-SCR-shRNA-PC-3 tumor and Lenti-SCR-shRNA-DU-145 tumor), at 4 wks after implantation of PC-3 cells and 7 wks after implantation of DU-145 cells, respectively. (B) Volumes of tumors with knockdown of hCtr1 (Lenti-hCtr1-shRNA-PC-3 and Lenti-hCtr1-shRNA-DU-45) were significantly smaller than those without knockdown of hCtr1 (Lenti-SCR-shRNA-PC-3 and Lenti-SCR-shRNA-DU-145), based on calculation from tumor size measured ex vivo with caliper. Error bar, SD.
DISCUSSION
Copper is required for cell proliferation, but excess copper is paradoxically cytotoxic. Copper homeostasis is tightly regulated by a delicate network of influx copper transporter (hCtr1), efflux copper transporters (ATP7A and ATP7B), copper chaperons (ATOX1, Cox17, CCS), and other copper binding molecules (26). In this study, we prepared polyclonal antibody specific for hCtr1 using recombinant hCtr1 protein expressed in Escherichia coli because the polyclonal hCtr1 antibody used previously (16) was not suitable for Western blot. Using newly prepared polyclonal antibody, we demonstrated elevated expression of hCtr1 in prostate cancer cells (PC-3, DU145, and C4-2) and immortalized prostate epithelial cells containing HPV oncogene (PZ-HPV-7, RWPE-1, RWPE-2) by Western blot, compared with hCtr1 expression in normal prostate epithelial cells (Fig. 1B). Previous detection of a low level of hCtr1 mRNA in PZ-HPV-7 cells by qRT-PCR in a prior study (27) might be due to variation of hCtr1 expression in the PZ-HPV-7 cells or a stability issue of the RNA samples used for that study. The reduction of cellular and tumor 64Cu uptake by RNAi-mediated knockdown of hCtr1 (Figs. 3 and 5) indicated that hCtr1 plays an important role in mediating the copper uptake required for uncontrolled proliferation of prostate cancer cells. Although hCtr1 expression in DU-145 cells was slightly higher than hCtr1 expression in PC-3 cells (Fig. 1), 64Cu uptake by tumors derived from DU-145 prostate cancer cells was found to be lower than 64Cu uptake by tumors derived from PC-3 cells (Figs. 3, 5, and 6). This difference might be related to variable expression of copper transporters, chaperons, or other copper carrier molecules, for example, metheothinine (28), between these 2 prostate cancer cells lines. Tumor uptake of 64Cu is expected to be variable based on the profile of various copper transporters, chaperons, and copper binding molecules, not simply depending on the expression level of hCtr1, supported by the findings from other investigators (29). Additional studies of the expression of hCtr1 and other copper transporters in the tumor tissue samples from the patients are warranted to determine their role in copper metabolism of prostate cancer. There has been increased interest in development of 64Cu radiopharmaceuticals for cancer imaging and therapy (30–32). Given expected variation of tumor 64Cu uptake, it is essential to set up appropriate controls to differentiate tumor uptake of free 64Cu ions disassociated from 64Cu-radiolabeled conjugates from tumor uptake of intact 64Cu-radiolabeled conjugates in the characterization of 64Cu-radiolabeled radiopharmaceuticals.
Prostate cancer is a heterogeneous disease characterized by variable genomic alterations, including different point mutations and chromosomal alterations such as deletions, insertions, or translocations (1,2). Given the complex heterogeneity of prostate cancer, the metabolism of prostate cancer may vary among different patients, even among different tumor lesions in the same individual patient diagnosed with prostate cancer. It is not surprising to have variable uptake of radiopharmaceuticals used for metabolic imaging of prostate cancer, including variable uptake of radiotracer 11C choline in prostate cancer (33). It is expected that copper metabolism of prostate cancer may vary in different patients, even in different lesions of prostate cancer in the same patient. Although it remains to be determined whether the use of 64CuCl2 PET/CT for screening or early diagnostic imaging of primary prostate cancer will be limited by the variability of copper metabolism, 64CuCl2 PET/CT might be useful for assessing copper metabolism of prostate cancer lesions detected by PET using a probe targeting a biomarker present on most prostate cancer cells, such as a radiopharmaceutical agent targeting prostate-specific membrane antigen (34), humanized radioiodinated minibody targeting prostate stem cell antigen (35), and radiolabeled peptide agent targeting vasoactive intestinal polypeptide receptor 1 (36). On the basis of requirement of copper for cell proliferation and tumor angiogenesis, and the recent finding that copper promotes invasion of the prostate cancer epithelial cells (37), it is postulated that copper metabolism status may be related to aggressiveness and metastasis of prostate cancer. Copper metabolism holds potential as a prognostic imaging biomarker in prostate cancer, using 64CuCl2 PET/CT. Safe usage of 64CuCl2 for the PET/CT imaging of human prostate cancer is supported by the prior use of 64CuCl2 for the assessment of copper metabolism in healthy human subjects and copper metabolism disorders in patients diagnosed with Wilson's disease without significant side effects (38,39); tiny amount of copper ions in a tracer dose of 64CuCl2 and reduction of Cu(II) ions for cellular uptake of Cu(I) ions mediated by hCtr1 (26), which should ease the concern of cytotoxicity of Cu(II) ions from 64CuCl2; and data from the recent radiation dosimetry of 64CuCl2 in a ATP7b−/− knockout mouse model of Wilson's disease, suggesting the safe usage of 64CuCl2 as a radioactive tracer for PET imaging in humans (24).
RNAi-mediated knockdown of hCtr1 was associated with the suppression of prostate cancer cell proliferation in vitro (Fig. 4). The molecular mechanism of suppression of cell proliferation by knockdown of hCtr1 remains to be illustrated, which may be related to a functional loss of some copper-requiring molecules regulating cell proliferation or abolishment of cell growth–stimulating activity of hCtr1 itself or disruption of its interaction with other molecules regulating cell proliferation. Knockdown of hCtr1 was associated with growth inhibition of prostate cancer xenografts in vivo (Fig. 7). We noted that growth of prostate cancer xenograft tumors derived from DU-145 cells was slower than growth of tumors derived from PC-3 prostate cancer cells (Fig. 7), although the level of hCtr1 expression in DU-145 cells was higher than the level of hCtr1 expression in PC-3 cells by Western blot (Fig. 1B). Differences in the growth of tumors derived from PC-3 and DU-145 cells may be related to the effects of other growth factors that are differentially expressed in PC-3 cells and enhance growth of tumors derived from PC-3 prostate cancers in vivo. The molecular mechanism of tumor growth inhibition by knockdown of hCtr1 remains to be elucidated, which may be related to reduction of cancer cell proliferation and suppression of tumor angiogenesis. It will be intriguing to study the effect of hCtr1 knockdown on cancer cell proliferation in vivo by PET/CT of tumor-bearing mice using 3′-deoxy-3′-18F-fluorothymidine (40), in conjunction with study of other molecules regulating cell proliferation.
CONCLUSION
Reduced tumor 64Cu uptake and tumor growth inhibition by RNAi-mediated knockdown of hCtr1 suggest that hCtr1 is a promising new theranostic target, which can be further developed for metabolic imaging of prostate cancer using 64CuCl2 PET/CT and personalized cancer therapy targeting copper metabolism.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This project was supported by the NIH (R21EB005331-01A2 and P30 CA142543). The production of 64Cu at Washington University School of Medicine was supported by the NIH/NCI (R24 CA86307). No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Saleh Ramesani for assistance with experimental PET/CT; Dennis J. Thiele, PhD, for pCDNA-3.1 vector encoding hCtr1; and Vinzenz M. Unger, PhD, for purified recombinant hCtr1 protein.
Footnotes
Published online Mar. 17, 2014.
- © 2014 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication August 27, 2013.
- Accepted for publication January 1, 2014.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Reduction in Copper Uptake and Inhibition of Prostate Cancer Cell Proliferation by Novel Steroid-based Compounds
- Development of Novel PSMA Ligands for Imaging and Therapy with Copper Isotopes
- Insights into Trace Metal Metabolism in Health and Disease from PET: "PET Metallomics"
- 64CuCl2 PET/CT in Prostate Cancer Relapse
- Assessment of Traumatic Brain Injury by Increased 64Cu Uptake on 64CuCl2 PET/CT
- Detection of Increased 64Cu Uptake by Human Copper Transporter 1 Gene Overexpression Using PET with 64CuCl2 in Human Breast Cancer Xenograft Model